
More Information
Submitted: 24 August 2019 | Approved: 30 August 2019 | Published: 02 September 2019
How to cite this article: Accurso V, Santoro M, Contrino AD, Casimiro P, Raso S, et al. Essential thrombocythemia: Biology, clinical features, thrombotic risk, therapeutic options and outcome. J Hematol Clin Res. 2019; 3: 053-059.
DOI: 10.29328/journal.jhcr.1001012
ORCID: orcid.org/0000-0002-0161-0957
Copyright License: © 2019 Accurso V, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Essential thrombocythemia: Biology, clinical features, thrombotic risk, therapeutic options and outcome
Vincenzo Accurso1*, Santoro M2, Contrino AD1, Casimiro P1, Raso S2, Carlisi M1, Sardo M1, Perez A3, Di Piazza F3, Russo A3 and Siragusa S1
1Hematology Division, University Hospital Policlinico “Paolo Giaccone”, Via del Vespro 129, 90127, Palermo, Italy
2Department of Surgical, Oncological and Stomatological Disciplines, University of Palermo, Via del Vespro 129, 90127, Palermo, Italy
3Department of Surgical, Oncological and Stomatological Disciplines, Section of Medical Oncology, University of Palermo, Via del Vespro 129, 90127, Palermo, Italy
*Address for Correspondence: Vincenzo Accurso, Hematology Division, University Hospital Policlinico “Paolo Giaccone”, Via del Vespro 129, 90127, Palermo, Italy, Tel: +393457901019; Email: [email protected]
Essential Thrombocythemia (ET) is currently classified as a Philadelphia negative myeloproliferative neoplasm (MPN) together with polycythemia vera (PV) and primary myelofibrosis (PMF); the latter can be further divided in pre-fibrotic primary myelofibrosis (pre-PMF) and overt myelofibrosis, as listed in the revised 2016 World Health Organization classification of myeloid malignancies (WHO 2016). Overall, respect to the others MPNs, ET is characterized by favorable prognosis, lower life expectancy if compared to the control population, increased risk of thrombohemorrhagic complications along with possible evolution in myelofibrosis and leukemic transformation. In this review the authors will review current knowledge on biology, clinical aspects, prognosis and stratification of thrombotic risk, therapeutic options and outcome in ET patients.
The revised 2016 World Health Organization classification for hematological malignancies and acute leukemia defines the criteria for the diagnosis of essential thrombocythemia. 4 major criteria and 1 minor criterion have been identified. Respectively, a platelet value greater than or equal to 459,000x109 L.; bone marrow biopsy showing proliferation mainly of the megakaryocyte lineage with increased number of enlarged, mature megakaryocytes with hyperlobulated nuclei; no significant increase or left shift in neutrophil granulopoiesis or erythropoiesis and very rarely (grade 1) increase in reticulin fibers; exclusion of WHO criteria for BCR-ABL CML, PV, PMF, myelodysplastic syndromes, or other myeloid neoplasms; presence of JAK2, CALR, or MPL mutations and a clonal marker or absence of evidence for reactive thrombocytosis. Diagnosis of ET requires meeting all major criteria or the first three major criteria and the minor criterion [1]. The estimated ET annual incidence in the United States is 2.5 cases per 100,000 whereas the prevalence is estimated to be 24 cases per 100,000. Furthermore, other studies estimated the annual ET incidence in Western countries in 0.2–2.5 cases per 100,000 with a prevalence of 38-57 cases per 100,000 [2,3]. Moreover, the incidence is higher in women than men population with an approximate ratio of 2:1 [4], the median age at diagnosis is 60 years with a 20% of patients receiving a diagnosis under 40 years of age. According to some studies over the last few years there has been a decrease in the age of ET diagnosis (56 years versus 60 years.) [5,6]. In our experience, in 234 consecutive patients diagnosed with ET between 1997 and 2019, 36.75% were diagnosed under 61 years of age, 26.07% under 51 years and 16.67% under 41 years. Interestingly, ET is characterized by overall favorable prognosis if compared to the other MPNs (but life-expectancy in ET is inferior to the control population) with an expected survival of 19.8 years (compared to 13.5 years in PV and 5.9 years in PMF). Cumulative incidence of blast transformation is lower in ET (3.8%) than in PV (6.8%) and PMF (14.2%). Moreover, similar cumulative incidence of fibrotic transformations is lower in ET (9.2%) than PV (21%) [7].
Diagnosis and mutational status
The remarkable knowledge acquired over the last 15 years in the field of molecular biology has improved the diagnostic ability of Philadelphia negative myeloproliferative neoplasms. V617F, janus kinase 2, located on chromosome 9p24, was first reported in 2005 and represents the most frequent mutation with an estimated frequency, in ET patients, of 50–60% [8-11]. Furthermore, in 2006 and 2013 further driver mutations have been identified, respectively, in MPL and CALR genes (calreticulin). MPL gene, myeloproliferative leukemia virus oncogene, is located on chromosome 1p34, also known as the thrombopoietin (TPO) receptor. Mutations on MPL are present in about 5% of patients with ET [12,13]. Finally, CALR gene, is located on chromosome 19p13.2 [14,15] and according to some authors, CALR mutations are closely related to platelets production. Unlike JAK2 and MPL mutations, CALR mutations are present in patients with thrombocytosis other than ET, such as PMF or refractory anemia with ring sideroblasts and marked thrombocytosis. The most frequent CALR mutations are, respectively, a 52 bp deletion (type 1) and a 5 bp insertion (type 2) [15,16]. In our series of 234 patients with ET, 71.79% showed V617F-JAK2 mutation, 8.9% CALR mutations, 1.71% MPL mutations [17], contrariwise, 52% of our ET patients are triple negative. Driver mutations were considered to be mutually exclusive. However, Mansier and colleagues have recently reported that additional mutations in CALR or MPL co-exist in approximately 10% of patients with low JAK2 burden. Unfortunately, the clinical significance of this double mutation is still unclear [17]. JAK2 mutated ET patients tend to be older, show higher hemoglobin levels and white blood cell counts as well as a lower platelet counts and serum EPO levels than patients without mutation [18]. Moreover, JAK2 mutated ET patients are more likely to develop thrombosis [19,20]. Furthermore, patients with CALR mutations have a different phenotype than V617F-JAK2 or MPL mutated patients. Indeed, they have a greater platelet value, a lower hemoglobin values and low leukocyte levels along with a lower thrombotic risk [21,22]. Several CALR variants, related to different phenotypes, have been identified. In particular, the type1-like variant is associated with a greater risk of myelofibrosis transformation, while the type2-like variant is associated with a more indolent clinical course [23]. The mutational landscape of MPNs is actually very complex. Indeed, alongside the aforementioned driver or phenotypic mutations (sufficient to determine the clinical phenotype of the disease), other sub-clonal mutations have been also identified. (TET2, ASXL1, CBL, IDH and IKZF1). Sub-clonal mutations can occur more often in conjunction with phenotypic mutations but can temporally either precede or follow them. To date, sub-clonal mutations do not have a clear diagnostic value, in fact they are present in myelodysplastic syndromes as well as in other hematological neoplasms such as acute leukemia. Interestingly, these mutations have been reported to have an important negative prognostic value in primary myelofibrosis (PMF). Their presence is associated with lower survival and a greater risk of acute leukemia evolution. Such mutations are identified in lower frequencies in ET (and PV) if compared to MF [24-27]. In 2013, the study reported by Nangalia et al. highlighted a median of 6.5 mutations in patients with ET if compared to 13 mutations found in patients with PMF. The most frequent sub-clonal mutations were found in DNMT3A, TET2, and ASXL1 [28]. Moreover, in a cohort of 181 ET patients, 46% were found to have somatic mutations including TET2 (13%), ASXL1 (11%), DNMT3A (6%), SF3B1 (5%), CEBPA (4%), along with mutations in TP53, SH2B3, EZH2, and CSF3R (each 2%). The impact on prognosis is not clear and today the use of next-generation sequencing in ET is still not routine neither recommended by guidelines [29].
Clinic feature
In MPN symptomatic burden is often severe and affect the majority of patients with the disease. Some authors have conducted an online survey on 1179 patients with MPN to quantify the burden of illness; results indicate that constitutional and splenomegaly associated symptoms were predominant (70% of patients) and compromised quality of life. Additional signs included fatigue (81%), pruritus (52%), night sweats (49%), bone pain (44%), fever (14%), and weight loss (13%). Most often patient’s experienced profound fatigue in excess of age matched controls and required assistance for daily activities or severe disabilities (34.5%) and reported MPN-associated disability (11.2%). Subsequently, the same authors attempted to validate a widely applicable 18-item tool (evaluation of the symptoms of myeloproliferative neoplasia Module [MPN-SAF]), coordinated with the Brief Fatigue Inventory, to evaluate the symptoms of myelofibrosis, essential thrombocythemia and polycythemia vera between potential cohorts in the United States, Sweden and Italy. Patients reported that symptoms associated with MPN disease were severe and frequent among all three MPNs. In a cohort of 161 ET patients the most frequently reported symptoms were fatigue (90.3%), numbness (58.8%), insomnia (58%), sad mod (57.3%), vertigo (56,1%), concentration problem (55.8%), inactivity (53.7%), early satiety (53.2%), night sweats (51.3%), sexual problem (51.0%), headache (47.1% ), abdominal discomfort (45.3%), bone pain (45.2%), cough (41.4%), itching (40.6%), abdominal pain (38.2%), weight loss (23.4%) and fever (17%). Patients with ET such symptoms present with a slightly lower frequency compared to PV and PMF and are significantly less severe [30]. In figure 1, we reported the symptoms and the relative frequency in a cohort of 234 ET patients enrolled at our institution.
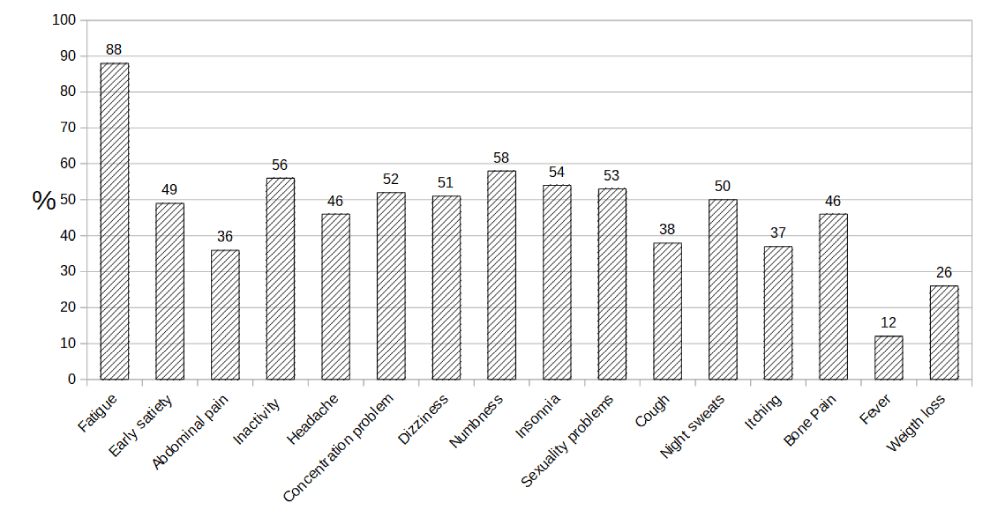
Figure 1: Frequency of symptoms in 234 ET patients.
Thrombotic risk and bleeding complication
An increased risk of vascular complications over time is the main clinical feature of ET. In a study conducted on 1,297 patients, thrombotic events, before or at the time of ET diagnosis, were reported in 231 cases (17.8%) [31]. MPNs are also a main causative factor of atypical site vein thrombosis, mainly in splanchnic (SVT) or cerebral veins [32]. In fact, about half of cases of Budd-Chiari syndrome and one-third of portal vein thrombosis are due to MPNs and SVT could be the first sign of the MPNs [33]. Therefore, several authors recommend the execution of JAK2, MPL or CARL mutational test in case of SVT [34-36]. The conventional thrombotic risk stratification in ET distinguishes patients in two risk groups: high risk group for patients older than 60 years or history of thrombosis, whereas the absence of both risk factors identifies the low risk group [37]. The classification of the thrombotic risk IPSETt contemplates the assignment of 2 points for previous thrombotic events in patients aged greater than 60 years and the presence of JAK2-V617F mutation, 1 point for age greater than 60 years and a point for the presence of cardiovascular risk factors (CV.R.). The low risk is defined by a score lower than 2, the intermediate risk by a score equal to 2 and the high risk by a score greater than 2 [38]. Recently, a revision of IPSET-thrombosis was achieved by the re-analysis of the original IPSET-thrombosis data set. The revised IPSET-Thrombosis uses three adverse variables to delineate four risk categories: age >than 60, thrombosis history and JAK2-V617F presence. Indeed, patients younger than 60 years old, negative history of thrombosis and no JAK2-V617F mutation are considered at very low risk; patients with JAK2-V617F mutation but no thrombosis history are considered at low risk; patients with JAK2-V617F mutation with a diagnosis of thrombosis are considered at intermediate risk; high risk category is defined by the presence of all three risk factors [39]. In 2017, further enhancement of IPSET-Thrombosis was proposed by Tefferi and Barbui adding the negative effect of MPL mutation [40]. Today, the current classification of thrombotic risk in patients with ET do not take into account CV.R and this condition does not currently influence the choice of cytoreductive therapy. However, our experience shows a close correlation between the presence of one or more cardiovascular risk factors (cigarette smoking, hypertension, diabetes, obesity, dyslipidemia) and thrombotic events. In fact, the frequency of thrombosis is lower in patients without cardiovascular risk factors (10/59) compared to patients with only one cardiovascular risk factor (18/92) and compared to patients with more than one cardiovascular risk factors (34/83) (p=0.0009). In table 1 we listed the cardiovascular risk factors in a cohort of 234 ET patients. The frequency of bleeding complications in patients with ET varies in different studies. In a 2012 international study conducted on 891 ET patients, 55 major bleeding events occurred (6.17%) [41]. previously, several reports indicated incidence of bleeding events in 36-37% of patients with ET. In 2005, Elliott and Teffery reported an incidence of bleeding events equal to 0.33% per person per year [42]. The most frequent hemorrhages concern the gastrointestinal and urogenital apparatus but may occur in several sites among which intracranial hemorrhages are particularly dangerous. Bleeding episodes are correlated with extreme thrombocytosis (platelet >1000-1500*109/L) and may be associated with acquired Von Willebrand disease (AVWS). The mechanism that determines the AVWS consists in the absorption of large Von Willebrand multimer by the platelet’s membranes. For these reasons, extreme thrombocytosis is an indication for cytoreductive therapy [43,44].
| Table 1: Cardiovascular risk factors in 234 ET patients. | ||
| Cardiovascular Risk | N° | % |
| Smoke | 31 | 13.30 |
| Hypertension | 148 | 63.52 |
| Obesity | 21 | 9.01 |
| Dyslipidemia | 56 | 24.03 |
| Diabetes | 33 | 14.16 |
Disease progression
In ET patient’s transformation to AML and/or post-ET myelofibrosis (post-ET MF) are rare events. Risk of transformation to acute leukemia was reported at 2–3% at 10 years and 5% at 15 years [45]. Subsequently, following the review of the case definition carried out by the WHO in 2016, the distinction between myelofibrosis in the pre-fibrotic phase and ET significantly reduced the incidence of transformation in post-ET myelofibrosis and acute leukemia [46]. When ET is confirmed on the basis of bone marrow morphology, the risk of leukemic transformation is estimated at 0.7% at 10 years and 2.1% at 15 years [47]. As for the evolution in myelofibrosis one of the largest longitudinal cohort studies reported the cumulative probability of myeloid disease transformation to be 3.8% at 10 years, 19.9% at 20 years, and 28.9% at 30 years [48]. But even in this case, the distinction between ET and pre-PMF could have an important impact on the incidence of transformation into myelofibrosis. Some risk factors seem to be related to leukemic transformation in patients with ET; the presence of anemia, platelets count <1000,000, advanced age and leukocytosis as well as reticulin grading and bone marrow cellularity [47,49] seem to play a pivotal role. Mutation subtype may play a role in the risk of transformation to AML or post-ET MF, but more data are still needed. For AML, while one study highlighted that JAK2-mutated patients have a higher risk of transformation to AML than CALR-mutated patients [50], these differences did not prove any statistical significance.
Therapeutic option
In 2018, the panel of experts from the European LeukemiaNet consortium (ELN) published the therapy recommendations for patients with MPNs. Based on the results of retrospective studies, the use of low-dose aspirin is recommended for high-risk patients according to the IPSET thrombosis classification. In low-risk or intermediate-risk patients aspirin is recommended at low doses if age> or =60 years and with uncontrolled cardiovascular risk factors or in the presence of JAK2 mutation [51]. Of particular interest is a study carried out on a cohort of 433 patients with low-risk ET in which the use of aspirin at low doses, in patients with a CALR mutation, seemed to increase the risk of bleeding without reducing the thrombotic risk [52]. Recently, a panel of Italian experts analyzed a series of unmet clinical needs in the management of the thrombotic risk and thrombotic events associated with Philadelphia-negative myeloproliferative neoplasms. The Panel agreed that optimization of LDA for primary prophylaxis of thrombosis in ET requires randomized trials investigating once daily versus more frequent LDA administrations to evaluate clinical outcomes in terms of thrombosis and bleeding [53]. For patients at high risk, the panel of European LeukemiaNet consortium, recommend the cytoreductive therapy and indicates hydroxyurea and rINFalfa as first line drugs. Even though the majority of the experts indicated anagrelide as an appropriate choice for first-line therapy in ET; the panel did not reach a consensus on recommending the agent in this setting, arguing that the evidence of non-inferiority with hydroxyurea was of insufficient quality, and the risk-benefit ratio unfavorable. Contrariwise, in patients with low or intermediate risk with well-controlled cardiovascular risk factors, cytoreductive therapy is not recommended. In case of intolerance or insufficient response to hydroxyurea therapy, the use of anegrelide or rINFalfa as a second-line therapy is recommended. The LeukemiaNet panel also recommends the early initiation of cytoreductive therapy in patients who switch to the high-risk category following the age of 60 year or following a major or hemorrhagic thrombotic event and/or a platelet count greater than 1,500.x 109 L. Cytoreduction may be also required for progressive myeloproliferation (e.g., increasing splenomegaly) or uncontrolled ET-related systemic symptoms [51].
Other drugs have been experimentally used in patients resistant or tolerant to hydroxyurea. A total of 18 patients in two sequential cohorts received an initial dose of 7.5mg or 9.4mg of imetelstat (Telomerase inhibitor) per kilogram of body weight intravenously once a week until attainment of a platelet count of approximately 250,000 - 300,000 per cubic millimeter. The primary end point was the best hematologic response. Imetelstat induced hematologic responses in all 18 patients, and 16 patients (89%) had a complete hematologic response. Molecular responses were seen in 7 of 8 patients who were positive for the JAK2-V617F mutation (88%; 95% confidence interval, 47 to 100). CALR and MPL mutant allele burden was also reduced by 15 to 66%. However, imetelstat therapy showed a high risk of grade 3-4 neutropenia, thrombotic and abnormal liver function test [54]. The histone deacetylase inhibitors vorinostat and givinostat were able to induce some signals of spleen reduction, symptoms relief and hematologic response in a substantial proportion of patient [55,56]. Verstovsek et al., used ruxolitinib in patients with ET resistant or intolerant to hydroxyurea. The results of this study demonstrated that patients can achieve clinically meaningful and durable reduction in platelet and WBC counts and improvements in ET-related symptoms with ruxolitinib treatment [57]. According to other authors, since the ET, especially after the revision of the case definition by the WHO in 2016, has a relatively benign course and the evolution in MF PET is quite a rare event, the use of Ruxolitinib appears questionable [58].
Pregnancy
Out of 234 patients with ET visited in our center, 33 of 161 women (20.49%) are fertile at the time of diagnosis, this implies the possibility to manage a pregnancy in this setting of ET patients. In this patients the risk of first-trimester fetal losses (about 3.5-fold) and placental complications (e.g. abruption, pre-eclampsia) is increased if compared to healthy women [59-61]. These events are possibly mediated by decidual thrombosis. Risk factors include previous pregnancy complications and possibly the presence of a JAK2-V617F mutation. Venous thrombosis may occur, particularly in the postpartum period, and the risk is higher in patients with a history of vascular events [62]. Furthermore, the incidence of postpartum hemorrhage is estimated at 9%. With regard to fetal complications, the perinatal mortality rate was 17 per 1000 live and stillbirths, and 22% were below the 10th percentile in weight, while 13% required admission to the neonatalcare unit [63]. In 2011 the panel of LeukemiaNet consortium defined treatment approach during pregnancy, although evidence about the impact on outcome is weak. For low risk patient low dose aspirin is recommended; moreover, prophylactic low molecular weight heparin dose after delivery, until 6 weeks postpartum, is recommended .For high risk patient with previous major thrombosis events or severe pregnancy complication a low molecular weight heparin dose throughout pregnancy is recommended (stop aspirin in case of bleeding complications; if platelet count >1,500×109/L: consider rINFalfa , if previous major bleeding: avoid aspirin and consider interferon alfa to reduce thrombocytosis) [64]. In 2014, consensus statement of the Haemostasis Working Party of the German Society of Hematology and Oncology (DGHO), the Austrian Society of Hematology and Oncology (€OGHO) and Society of Thrombosis and Haemostasis Research, offer similar recommendations [65].
Survival
Overall, ET is considered as an indolent disease. The 15-year survival is 75%, with risk factors for poor survival identified as lower hemoglobin level than normal, age 60, leukocyte count ≥15×109/L smoking, diabetes, thrombosis, thrombocythemia, and male gender [66]. The International Prognostic Score for ET (IPSET) is a prognostic model that was developed to predict survival at diagnosis. Patients identified as intermediate risk (total score 1–2) had a median survival of 24.5 years, while patients identified as high risk (total score 3–4) had a median survival of 13.8 years [67].
Familial ET
Rare cases of ET in the same household are reported. Predisposition haplotypes, including 46/1, [68] and predisposition alleles have been identified and seem to confer an increased risk of developing not only MPN but also JAK2-V617F clonal hematopoiesis [69-71.] Maffioli, Mora et al. analyzed 22 cases of familial MPNs, for a total of 45 patients. Within this cohort of 22 familial MPN pedigrees disease phenotype and genotype are heterogeneously distributed and occurrence is more frequently vertical [72]. Among our patients we have identified a unique case of familial ET. The case concerns a brother and a sister who have in common only one parent (the father); in both cases the diagnosis of ET has been done according to the WHO 2016 criteria, of particular interest is the presence of a CALR mutation in the male and a JAK2-V617F mutation in the sister (unpublished data). Unfortunately, it has not been possible to perform further molecular testing to the parent as he has been dead for several years. We recommend that physicians interview all patients with ET to determine whether there is a family history of thrombocytosis or MPN [73].
ET is a relatively rare chronic myeloproliferative disease, characterized by a favorable prognosis with similar or slightly lower survival than the general population. The distinction made in 2016 by the WHO in pre-PMF and myelofibrosis overt has further improved the prognosis as several cases of ET have been reclassified to pre-PMF which has a slightly worse prognosis. The mutational state and the thrombotic risk may influence the therapeutic choice. The knowledge acquired in the last two decades, especially in terms of molecular biology, have greatly improved understanding of the pathogenesis and the ability to diagnose negative Philadelphia myeloproliferative diseases. To date, however, still several cases of ET are diagnosed after the thrombotic event. Therefore, early diagnosis could further minimize the risk of thrombotic even and improve the outcome.
- Daniel AA, Attilio Orazi, Robert Hasserjian, Thiele J, Borowitz MJ, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. BLOOD. 2016; 127: 20. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27069254
- Ma X, Vanasse G, Cartmel B, Wang Y, Selinger HA. Prevalence of polycythemia vera and essential thrombocythemia. Am J Hematol. 2008; 83: 359-362. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18181200
- Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006; 32: 171-173. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16673273
- Cortelazzo S, Viero P, Finazzi G, D'Emilio A, Rodeghiero F, et al. Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol. 1990; 8: 556-562. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2307991
- Fenaux P, Simon M, Caulier MT, Lai JL, Goudemand J, et al. Clinical course of essential thrombocythemia in 147 cases. Cancer 1990; 66: 549-556. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2364366
- Tefferi A, Fonseca R, Pereira DL, Hoagland HC. A long-term retrospective study of young women with essential thrombocythemia. Mayo Clin Proc. 2001; 76: 22-28. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11155408
- Tefferi A, Barbui T. Polycytemia vera and essential thrombocythemia: 2019 update on diagnosis, risk stratification and management. Am J Hematol. 2019; 94: 133-143. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30281843
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365: 1054-1061. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15781101
- Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia M with myelofibrosis. Cancer Cell. 2005; 7: 387-397.
- James C, Ugo V, Le Couedic J-P, Staerk J, Delhommeau F, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005; 434: 1144-1148. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15793561
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, et al. A gain-offunctionmutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005; 352: 1779-1790. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15858187
- Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, et al. Mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006; 108: 3472-3476. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16868251
- Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006; 3: e270. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16834459
- Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin:the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood. 2014, 123: 3714-3719. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24786775
- Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013; 369: 2379-2392.
- Marty C, Pecquet C, Nivarthi H, El-Khoury M1, Chachoua I, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016; 127: 1317-1324. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26608331
- Mansier O, Luque Paz D, Ianotto JC, Le Bris Y, Chauveau A, et al. Clinical and biological characterization of MPN patients harboring two driver mutations, a French intergroup of myeloproliferative neoplasms (FIM) study. Am J Hematol. 2018; 93: 84-86. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29266414
- Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005; 366: 1945-1953. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16325696
- Cheung B, Radia D, Pantelidis P, Yadegarfar G, Harrison C. The presence of the JAK2 mutation is associated with a higher haemoglobin and increased risk of thrombosis in essential thrombocythaemia. Br J Haematol. 2006; 132: 244-245. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16398659
- Zhang S, Qiu H, Fischer BS. JAK2 V617F patients with essential thrombocythemia present with clinical features of polycythemia vera. Leuk Lymphoma. 2008; 49: 696-699. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18398736
- Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, et al. CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014; 123: 1544-1551. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24366362
- Rotunno G, Mannarelli C, Guglielmelli P, Pacilli A, Pancrazzi A, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014; 123: 1552-1555. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24371211
- Pietra D, Rumi E, Ferretti VV, Di Buduo CA, Milanesi C, et al. Differential clinical effects of different mutation subtypes in CALR mutant myeloproliferative neoplasms. Leukemia. 2016; 30: 431-438. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26449662
- Cabagnols X, Favale F, Pasquier F, Messaoudi K, Defour JP, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. 2016; 127: 333-342. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26450985
- Milosevic Feenstra JD, Nivarthi H, Gisslinger H, Leroy E, Rumi E, et al. Sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016; 127: 325-332. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26423830
- Yamamoto Y, Iba S, Abe A, et al. Elongation of MPL transmembrane domain is a novel activating-mutation in essential thrombocythemia. Blood. 2015; 126: 1628-1628.
- Tefferi A. Novel mutations and their functional andclinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010; 24: 1128-1138. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20428194
- Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013; 369: 2391-2405. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24325359
- Tefferi A, Lasho TL, Finke C. Targeted next-generation sequencing in polycythemia vera and essential thrombocythemia. Blood. 2015; 126: 354-354.
- Scherber R, Dueck AC, Johansson P, Barbui T, Barosi G, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): International Prospective Validation and Reliability Trial in 402 patients. Blood. 2011; 118: 2. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21536863
- Andriani A, Latagliata R, Anaclerico B, Spadea A, Rago A, et al. Spleen enlargement is a risk factor for thrombosis in essential thrombocythemia: Evaluation on 1,297 patients. American Journal of Hematology. 2016; 91: 3. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26748894
- Dentali F, Ageno W, Rumi E, Casetti I, Poli D, et al. Cerebral venous thrombosis and myeloproliferative neoplasms: results from two large databases. Thromb Res. 2014; 134: 41-43. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24787989
- Chait Y, Condat B, Cazals-Hatem D, Rufat P, Atmani S, et al. Relevance of the criteria commonly used to diagnose myeloproliferative disorder in patients with splanchnic vein thrombosis. Br J Haematol. 2005; 129: 553-560. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15877740
- Rumi E, Cazzola M. How I treat essential thrombocythemia. Blood. 2016; 128: 2403-2414. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27561316
- Dentali F, Squizzato A, Brivio L, Appio L, Campiotti L, et al. JAK2V617F mutation for the early diagnosis of Ph myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood. 2009; 113: 5617-5623. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19273837
- Roques M, Park JH, Minello A, Bastie JN, Girodon F. Detection of the CALR mutation in the diagnosis of splanchnic vein thrombosis. Br J Haematol. 2015; 169: 601-603. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25413838
- Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, et al. Philadelphia – Negative classical myeloproliferative neoplasms: critical concepts and management recomandation from European LeukemiaNet. J Clin Oncol. 2011; 29: 761-770. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21205761
- Barbui T, Finazzi G, Carobbio A, et al. Development and validation an international Prognostic Score of thrombosis in World Health Organization essential thrombo cythemia. Blood. 2012; 120: 5128-5133. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23033268
- Haider M, Gangat N, Lasho T, Abou Hussein AK, Elala YC, et al. Validation of the revised international prognostic score of thrombosis for essential thrombocythemia (IPSET-thrombosis) in 585 Mayo clinic patients. Am J Hematol. 2016; 91: 4. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26799697
- Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2017 update, risk stratification, and management. Am J Hematol. 2017; 92: 94. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27991718
- Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, et al. Incidence and risk factors for bleeding in 1104 patients with essential thrombocythemia or prefibrotic myelofibrosis diagnosed according to the 2008 WHO criteria. Leukemia. 2012; 26: 716-719. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21926959
- Elliott MA, Tefferi A. Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol. 2005; 128: 275-290. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15667529
- Budde U, van Genderen PJ. Acquired von Willebrand disease in patients with high platelet counts. Semin Thromb Hemost. 1997; 23: 425-431. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9387201
- Federici AB, Rand JH, Bucciarelli P, Budde U, Van Genderen PJ, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. 2000; 84: 345-349. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10959711
- Passamonti F, Rumi E, Arcaini L, Boveri E, Elena C, et al. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica. 2008; 93: 1645-1651. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18790799
- Gangat N, Wolanskyj AP, McClure RF, Li CY, Schwager S, et al. Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia. 2006; 21: 270-276. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17170720
- Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011; 29: 3179-3184. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21747083
- Wolanskyj AP, Schwager SM, McClure RF, Larson DR, Tefferi A. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc. 2006; 81: 159-166. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16471068
- Abdulkarim K, Ridell B, Johansson P, Kutti J, Safai-Kutti S, et al. The impact of peripheral blood values and bone marrowfindings on prognosis for patients with essential thrombocythemia and polycythemia vera. Eur J Haematol. 2011; 86: 148-155. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21059102
- Pietra D, Rumi E, Ferretti VV, Di Buduo CA, Milanesi C, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia. 2016; 30: 431-438. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26449662
- Barbui T, Tefferi A, Vannucchi AM, Passamonti F, Silver RT, et al, Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018; 32: 1057-1069. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29515238
- Alvarez-Larran A, Pereira A, Guglielmelli P, Hernández-Boluda JC, Arellano-Rodrigo E, et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with CALR mutation. Haematologica. 2016; 101: 926-931. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27175028
- Barbui T, De Stefano V. Addressing and proposing solutions for unmet clinical needs in the managementof myeloproliferative neoplasm-as-associated thrombosis: A consensus-based position paper. Blood Cancer Journal. 2019; 9: 61.
- Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, Spitzer G, Odenike O, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015; 373: 920-928. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26332546
- Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, et al. A pilot study of the histonedeacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010; 150: 446-455. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20560970
- Andersen CL, McMullin MF, Ejerblad E, Zweegman S, Harrison C, et al. A phase II study of vorinostat (MK-0683) in patients with polycythaemia vera and essential thrombocythaemia. Br J Haematol. 2013; 162: 498-508. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23758082
- Verstovsek S, Passamonti F, Rambaldi A, Barosi G5, Rumi E, et al. Ruxolitinib for essential thrombocythemia refractory to or intolerant of hydroxyurea: long-term phase 2 study results. Blood. 2017; 130: 1768-1771. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28827411
- Mora B, Passamonti F. Developments in diagnosis and treatment of essential thrombocythemia. Expert Review of Hematology. 2019; 12: 159-171. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30793984
- Wright CA, Tefferi A. A single institutional experience with 43 pregnancies in essential thrombocythemia. Eur J Haematol. 2001; 66: 152-159. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11350483
- Lavi N, Brenner B, Avivi I. Management of pregnant women with myeloproliferative neoplasms. Thromb Res. 2013; 131: S11-S13. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23452731
- Passamonti F, Randi ML, Rumi E, Pungolino E, Elena C, et al. Increased risk of pregnancy complications in patients with essential thrombocythemia carrying the JAK2 (617VF) mutation. Blood. 2007; 110: 485-489. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17426257
- Randi ML, Bertozzi I, Rumi E, Elena C, Finazzi G, et al. Pregnancy complications predict thrombotic events in young women with essential thrombocythemia. Am J Hematol. 2014; 89: 306-309. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24265194
- Alimam S, Bewley S, Chappell LC, Knight M, Seed P, et al. Pregnancy outcomes in myeloproliferative neoplasms: UK prospective cohort study. Br J Haematol. 2016; 175: 31-36. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27612319
- Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, et al. Philadelphia negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European leukemianet. J Clin Oncol. 2011; 29: 761-770. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21205761
- Kreher S, Ochsenreither S, Trappe RU. Prophylaxis and management of venous thromboembolism in patients with myeloproliferative neoplasms: consensus statement of the Haemostasis Working Party of the German Society of Hematology and Oncology (DGHO),the Austrian Socie-ty of Hematology and Oncology (€OGHO) and Society of Thrombosis and Haemostasis Research (GTH eV). Ann Hematol. 2014; 93: 1953-1963.
- Passamonti F, Rumi E, Pungolino E, Malabarba L, Bertazzoni P, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004; 117: 755-761. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15541325
- Passamonti F, Thiele J, Girodon F, Rumi E, Carobbio A, et al. A prognostic model to predict survival in 867 World Health Organization–defined essential thrombocythemia at diagnosis: a study by the International Working Group on Myelofibrosis Research and Treatment. Blood. 2012; 120: 1197-1201. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22740446
- Napolitano M, Siragusa S. Comparison of clinical and laboratory data, including JAK-2 46/1 haplotipe, between patients With Idiopathic Erythrocytosis and Polycythemia Vera. EHA22 Abstract book PB 2039.
- Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood. 2016; 128: 1121-1128. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27365426
- Rumi E, Passamonti F, Pietra D, Della Porta MG, Arcaini L, et al. JAK2 (V617F) as an acquired somatic mutation and a secondary genetic event associated with disease progression in familial myeloproliferative disorders. Cancer. 2000; 107: 2206–2211. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16998940
- Bellanne-Chantelot C, Chaumarel I, Labopin M, Labopin M, Bellanger F, et al. Genetic and clinical implications of the Val617Phe JAK2 mutation in 72 families with myeloproliferative disorders. Blood. 2006; 108: 346-352.
- Maffioli M, Mora B. Familial myeloproliferative neoplasms. A single institution: analysis of 22 families. EHA23 Abstract Book PS 1361.
- Rumi E, Cazzola M. How we treat essential thrombocythemia. Blood. 2016; 128: 2403-2414. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27561316