
More Information
Submitted: November 13, 2024 | Approved: November 19, 2024 | Published: November 20, 2024
How to cite this article: Delicou S, Moraki M, Papatheodorou E, Xydaki A. Treatment Options for Congenital Dyserythropoietic Anemias (CDAs): Advances in Bone Marrow Transplantation, Gene Therapy, and Targeted Therapies. J Hematol Clin Res. 2024; 8: 027-033. Available from:
?https://dx.doi.org/10.29328/journal.jhcr.1001031.
DOI: 10.29328/journal.jhcr.1001031
Copyright License: © 2024 Delicou S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Personalized interventions, Congenital Dyserythropoietic Anaemia (CDA), Red blood cell maturation, Targeted therapies

Treatment Options for Congenital Dyserythropoietic Anemias (CDAs): Advances in Bone Marrow Transplantation, Gene Therapy, and Targeted Therapies
Sophia Delicou1*, Maria Moraki2, Elena Papatheodorou3 and Aikaterini Xydaki4
1Hematologist, Thalassemia and Sickle Cell Unit, Expertise Center of Hemoglobinopathies and Their Complications, Hippokrateio General Hospital Athens, Greece
2Pediatrician, Thalassemia and Sickle Cell Unit, Expertise Center of Hemoglobinopathies and Their Complications, Hippokrateio General Hospital Athens, Greece
3Cardiology Resident, Thalassemia and Sickle Cell Unit, Expertise Center of Hemoglobinopathies and Their Complications, Hippokrateio General Hospital Athens, Greece
4Internist, Thalassemia and Sickle Cell Unit, Expertise Center of Hemoglobinopathies and Their Complications, Hippokrateio General Hospital Athens, Greece
*Address for Correspondence: Sophia Delicou, MD, Hematologist, Thalassemia and Sickle Cell Unit, Expertise Center of Hemoglobinopathies and Their Complications, Hippokrateio General Hospital Athens, Greece, Email: [email protected]
Congenital Dyserythropoietic Anaemia (CDA) is a rare genetic disorder that affects the maturation of red blood cells. The disorder is classified into different types, with a prevalence ranging from 1 in 100,000 to 1 in 1,000,000 individuals. Treatment strategies are designed with the primary focus on symptom management, the prevention and treatment of complications, and the underlying disease pathophysiology. The advent of bone marrow transplantation, gene therapy, and targeted therapies has considerably expanded the scope for therapeutic intervention in CDAs. Supportive care, including blood transfusions and iron chelation therapy, has demonstrated efficacy in managing iron overload and improving overall survival rates. The potential of gene therapy, targeted therapies, and hematopoietic growth factors in the treatment of CDA is currently being investigated. Further research and clinical trials are required to develop more effective and personalized therapeutic interventions.
Congenital Dyserythropoietic Anemia (CDA) is a rare genetic disorder that affects the normal development of red blood cells. It is classified into different types based on its genetic and clinical characteristics. CDA Type I is caused by mutations in the CDAN1 and C15orf41 genes and is characterized by macrocytic anemia and congenital abnormalities. CDA Type II, the most common form, is caused by mutations in the SEC23B gene and is characterized by normocytic anemia and elevated erythroferrone levels. CDA Type III is the rarest form and is caused by a mutation in the KIF23 gene, resulting in absent or moderate anemia and giant multinucleated erythroblasts. CDA Type IV is caused by a mutation in the KLF1 gene and is characterized by severe hemolytic anemia with cytoplasmic inclusions [1,2].
The prevalence of CDA is estimated to be between 1 in 100,000 and 1 in 1,000,000 individuals, and it can occur at any age. The rarity and diversity of the disease often lead to underdiagnosis, highlighting the need for improved diagnostic techniques and research [1,2].
Diagnosis of CDA involves a combination of hematological, biochemical, and morphological evaluations. These include complete blood count, peripheral blood smear, bone marrow aspiration and biopsy, iron studies, and tests for liver function. Molecular diagnosis using next-generation sequencing has significantly improved the accuracy and speed of diagnosis [3].
The management of Congenital Dyserythropoietic Anemias (CDAs) is complex and multifaceted, tailored to the severity of the disease, the specific CDA subtype, and the individual patient’s clinical presentation. Treatment strategies are primarily directed towards symptom alleviation, complication management, and addressing the underlying disease pathophysiology. Recent advances in bone marrow transplantation, gene therapy, and targeted therapies have significantly broadened the therapeutic landscape for CDAs.
This discussion elucidates these advanced modalities, incorporating detailed insights from recent literature.
Supportive care
Supportive care remains the cornerstone of CDA management, especially for patients with mild to moderate disease severity.
Transfusions: Regular blood transfusions are often necessary to manage severe anemia and maintain adequate hemoglobin levels. However, frequent transfusions pose a risk of iron overload, necessitating meticulous monitoring and management. Transfusions provide immediate amelioration of anemia but contribute to iron accumulation, requiring adjustments to the transfusion regimen based on ongoing iron level assessments [1,3,4]
Iron chelation therapy: For patients undergoing frequent transfusions, iron chelation therapy is crucial to prevent iron overload and subsequent organ damage [5]. Deferasirox and deferoxamine are the primary agents used, with deferasirox offering the convenience of oral administration, whereas deferoxamine is administered via infusion [6,7]. Effective iron chelation has been demonstrated to significantly mitigate the risks of iron overload-related complications, such as hepatic and cardiac dysfunction. Iron chelation therapy has demonstrated significant effectiveness in managing iron overload, as evidenced by several key findings: A reduction in serum ferritin levels is evident in patients undergoing treatment. The mean decrease in serum ferritin levels observed in patients treated with deferasirox was approximately 500 ng/mL over a 12-month period [6,7]
Additionally, improvements in organ function were observed. Long-term therapy has been associated with enhanced liver function and a 50% reduction in the likelihood of developing liver cirrhosis among patients who have undergone effective chelation [8,9].
The data presented thus far have been derived from clinical trials. Deferasirox was observed to reduce liver iron concentration by 30% in patients with thalassemia after two years, confirming its efficacy in the management of iron overload. Adherence to oral chelation therapy was significantly higher (70%) in comparison to injectable therapies, resulting in superior outcomes in iron management. Patients who were adherent to iron chelation therapy exhibited a 40% reduction in the risk of developing heart failure and a 30% reduction in the risk of developing diabetes when compared to non-adherent patients. These findings collectively highlight the beneficial impact of iron chelation therapy on patient health and the management of iron overload [8,9].
Bone Marrow Transplantation (BMT)
BMT is currently the only curative treatment for severe cases of CDAs, particularly CDA types I and II [10,11].
BMT is considered for patients with severe anemia unresponsive to supportive care or those with substantial disease complications. The decision to pursue transplantation is influenced by the patient’s age, overall health, and availability of a compatible donor. BMT is especially indicated for patients facing life-threatening complications or experiencing a severely diminished quality of life due to frequent transfusions. Recent advancements in BMT techniques [12] have notably improved patient outcomes. The adoption of reduced-intensity conditioning (RIC) regimens has expanded BMT accessibility to older patients and those with comorbidities who may not tolerate traditional myeloablative conditioning [13]. Enhanced graft-versus-host disease (GVHD) prophylaxis and post-transplant care have reduced GVHD incidence and severity, contributing to improved survival rates. Innovations in donor matching and stem cell sourcing, such as the utilization of haploidentical donors and umbilical cord blood, have broadened the donor pool, increasing the likelihood of finding suitable matches.
The following section presents a detailed analysis of the specific statistics regarding the efficiency of Bone Marrow Transplantation (BMT) in Congenital Dyserythropoietic Anaemia (CDA).
The overall survival rate for patients with CDA who undergo bone marrow transplantation is estimated to be 70% to 90% in cases where a matched sibling is a donor. These outcomes significantly improve the prognosis for these patients. Engraftment rates for CDA patients receiving BMT are generally high, with studies indicating rates of approximately 80% to 90%. Neutrophil and platelet engraftment usually occurs within approximately 20 to 30 days following the transplant procedure. Following a successful bone marrow transplant, studies have indicated that over 70% of patients with CDA become transfusion-independent, indicating a significant reduction in the need for blood transfusions after the procedure. The incidence of Graft-Versus-Host Disease (GVHD) in CDA patients undergoing BMT has been reported to occur in approximately 30% to 50% of cases. The severity of GVHD is dependent on the compatibility between donor and recipient and the conditioning regimens employed. Long-term studies have demonstrated that patients who undergo successful BMT for CDA report significant improvements in quality of life, with many returning to their pre-transplant level of activity and experiencing fewer health complications related to their underlying conditions [13,14]. Studies have confirmed that BMT improves long-term outcomes for CDA patients, significantly reducing transfusion dependency and enhancing overall quality of life.
Gene therapy
Gene therapy is emerging as an increasingly promising approach for CDAs, particularly those with identifiable genetic mutations [15].
The objective of gene therapy is the correction of the underlying genetic mutations that are responsible for the disease. The development of techniques, such as the CRISPR/Cas9 gene editing approach [16], has the potential to enable the targeting of specific mutations in CDA-associated genes. This approach requires the editing of the patient’s cells to rectify the underlying genetic defect, which is then reintroduced into the patient’s body to facilitate the production of healthy erythrocytes.
The potential efficacy of gene therapy in restoring normal erythropoiesis has been substantiated in preclinical models [17]. For instance, the correction of mutations in the SEC23B gene in CDAII models has been demonstrated to result in significant improvements in erythroid differentiation and function. These studies, conducted in vitro and in animal models, have played an essential role in validating the potential of gene therapy before advancing to human trials [18].
Ongoing clinical trials are assessing the safety and efficacy of gene therapy in patients with various inherited anemias, including CDAs. Initial results are promising, indicating significant improvements in erythropoiesis and reduction in disease symptoms. However, further studies are essential to confirm long-term outcomes and safety profiles.
Recent progress in gene therapy techniques, particularly the development of lentiviral vectors and CRISPR/Cas9 technology, has enhanced the potential for therapeutic applications.
CRISPR/Cas9 represents a revolutionary advance in the field of gene editing, offering the potential for highly precise alterations to DNA in both in vitro and in vivo settings [19]. In the area of Congenital Dyserythropoietic Anaemia (CDA), clinical trials are investigating the potential of this technology to rectify specific genetic mutations that are responsible for the disease. The principal objectives of these studies are twofold: firstly, to ascertain whether the gene editing process can be undertaken without any inadvertent impact on other regions of the genome (off-target effects); and secondly, to evaluate whether the corrected genes can restore normal functionality in haematopoietic stem cells, which are essential for red blood cell production. The potential for successful gene editing is that it could enhance erythropoiesis, the production of red blood cells, and help alleviate the symptoms of anaemia in patients affected by the disease.
Lentiviral vectors [20,21], derived from lentiviruses, function as vehicles for the delivery of therapeutic genes into cells. In the context of CDA, clinical trials are utilizing these lentiviral vectors as a vehicle for the introduction of genes with the objective of boosting red blood cell production. The main areas of focus for these trials are as follows: firstly, confirmation of the ability of lentiviral vectors to effectively transfer therapeutic genes into hematopoietic stem cells thereby improving red blood cell counts and alleviating the symptoms of anemia. Current trials suggest that gene therapy holds significant promise for achieving long-lasting improvements in erythropoiesis and reducing disease-related complications in CDA.
Targeted therapies
Understanding the molecular mechanisms underlying CDAs has facilitated the development of targeted therapies. Preliminary results from clinical trials indicate that targeted therapies, particularly activin receptor ligand traps, significantly improve hematologic parameters and reduce transfusion needs in CDA patients.
Erythroferrone modulation: Erythroferrone (ERFE) is a fundamental hormone that exerts a substantial influence on iron metabolism, particularly in the context of anemia and associated conditions such as Congenital Dyserythropoietic Anemia (CDA). The following section provides an overview of its function, modulation, and therapeutic implications.
Erythroferrone (ERFE) is produced by erythroid cells, which are the precursors to red blood cells, following the onset of stimuli that promote erythropoiesis, including conditions characterised by anemia (a reduction in red blood cells) or hypoxia (insufficient oxygen levels) [22,23].
The primary role of ERFE is the inhibition of hepcidin, a hormone derived from the liver that is responsible for regulating iron balance. By suppressing hepcidin, ERFE enhances the availability of iron necessary for hemoglobin synthesis in red blood cells, which is essential for effective erythropoiesis. In CDA, the production of red blood cells is ineffective, resulting in a shortage of healthy red blood cells despite increased erythropoietic activity. This often results in elevated ERFE levels. The excessive production of ERFE in CDA causes significant suppression of hepcidin, which in turn leads to increased dietary iron absorption and mobilization from body stores [23,24]. This dysregulation can result in iron overload, characterized by the accumulation of excess iron in organs, which may potentially cause damage to the liver, heart, and other tissues.
As ERFE plays a crucial role in iron metabolism and the pathophysiology of CDA, targeting ERFE or its signaling pathways represents a promising therapeutic strategy. Potential therapeutic approaches may include:
1. Agents to modulate ERFE levels: These could be pharmacological agents that reduce ERFE levels in CDA patients, to restore normal hepcidin regulation and iron homeostasis [25,26].
2. Blocking ERFE effects: Alternatively, therapies could focus on inhibiting the effects of ERFE on hepcidin secretion, thereby reducing iron overload and enhancing erythropoiesis [25,26].
Additional trials are investigating prospective pharmaceutical agents that can either regulate ERFE levels or mimic its action, with the aim of enhancing erythropoiesis and alleviating iron overload in CDA patients. Some of these pharmaceutical agents may target the TGF-β signaling pathway, which is known to regulate ERFE. ERFE-targeted therapies represent a promising strategy to address iron overload in CDA, with ongoing studies supporting their potential to improve clinical outcomes.
Activin receptor ligand traps: It has been demonstrated that activin receptor ligand traps, such as sotatercept (ACE-011) and luspatercept (ACE-536), have the potential to be highly efficacious therapeutic agents in the management of Congenital Dyserythropoietic Anaemia (CDA) and other forms of ineffective erythropoiesis. These agents act by inhibiting the signalling pathways of the transforming growth factor-beta (TGF-β) superfamily, which includes activins and Growth Differentiation Factors (GDFs) that exert a negative regulatory effect on erythropoiesis.
Inhibition of these negative regulators may thus promote late-stage erythropoiesis and improve red blood cell production in conditions characterised by ineffective erythropoiesis, such as CDA [2,27].
The capacity of these agents to neutralise the effects of negative regulators of erythropoiesis suggests that they could potentially enhance haemoglobin levels and reduce the necessity for blood transfusions in patients with CDA.
Clinical trials: A number of clinical trials are currently in progress, investigating the efficacy and safety of activin receptor ligand traps in patients with CDA and related anemias. Notable ongoing trials are outlined below:
Sotatercept (ACE-011): This study will evaluate the efficacy of sotatercept in improving haemoglobin levels and reducing transfusion requirements in patients with CDA.
Ongoing trials are assessing the safety and efficacy of sotatercept in various populations, including those with beta-thalassaemia and CDA [28,29].
Luspatercept (ACE-536): Clinical trials are currently being conducted with the objective of evaluating the safety and efficacy of luspatercept in patients with various forms of anaemia, including CDA [30,31].
Hematopoietic growth factors: In select cases, erythropoiesis-stimulating agents (ESAs), such as epoetin alfa, may be considered as a means of stimulating red blood cell production. There is considerable variation in the efficacy of ESAs in CDAs, with outcomes dependent on the specific subtype and underlying pathophysiology. Erythropoiesis-stimulating agents (ESAs) are typically used to enhance short-term erythropoiesis, particularly in patients who have not responded to other treatments. This section provides a structured overview of the hematopoietic growth factors relevant to Congenital Dyserythropoietic Anemia (CDA), including their functions, relevance to CDA, and ongoing clinical trials for each factor [2,32,33].
1. Erythropoietin (EPO): EPO is a glycoprotein hormone primarily produced by the kidneys. It stimulates the production of Red Blood Cells (RBCs) in the bone marrow, thus improving haemoglobin levels and reducing the need for blood transfusions. Erythropoiesis-Stimulating Agents (ESAs), such as epoetin alfa, may be considered as a means of stimulating red blood cell production [34]. These agents act by mimicking the action of endogenous erythropoietin, which is primarily produced in the kidneys and plays a crucial role in promoting erythroid progenitor cell proliferation and differentiation in the bone marrow. While ESAs have shown variable efficacy in Congenital Dyserythropoietic Anemias (CDAs), they are particularly beneficial in cases where residual erythropoietic capacity can be leveraged to improve hemoglobin levels. Clinical trials have demonstrated modest improvements in anemia-related symptoms, though the response largely depends on the specific CDA subtype and its underlying pathophysiology. However, its efficacy may be limited in CDA due to the disease’s underlying mechanisms, which involve ineffective erythropoiesis.
2. Granulocyte-Macrophage: Colony-Stimulating Factor (GM-CSF) GM-CSF stimulates the production of granulocytes and macrophages from the bone marrow and can enhance erythropoiesis through an indirect mechanism. GM-CSF may improve overall haematopoietic function, potentially benefiting patients with CDA by enhancing the production of blood cells. Research suggests that GM-CSF may have a supportive role in improving haematopoietic function, although specific studies in CDA are limited.
3. Thrombopoietin (TPO): TPO is a key regulator of platelet production and also influences erythropoiesis. TPO may help to enhance the overall haematopoietic environment, potentially benefiting patients with CDA by supporting red blood cell production. The role of TPO in enhancing erythropoiesis in CDA is still being evaluated, with additional studies needed to clarify its effectiveness.
Hematopoietic growth factors such as EPO, GM-CSF, and TPO hold promise in the management of congenital dyserythropoietic anemia by stimulating erythropoiesis and improving overall hematopoietic function. Ongoing clinical trials are crucial for determining their efficacy and safety in this specific patient population, and the results will provide valuable insights into their potential roles in comprehensive CDA treatment strategies.
Combination therapies
Several ongoing trials are exploring the potential of combining activin receptor ligand traps with other therapies to enhance their efficacy in treating CDA.
Ongoing studies are investigating the potential for enhanced efficacy through the combination of these agents with other erythropoiesis-stimulating therapies [35].
The use of activin receptor ligand traps represents a novel approach to the management of congenital dyserythropoietic anemia, with the agents targeting the negative regulators of erythropoiesis [36]. The efficacy and safety of this therapeutic strategy for improving hemoglobin levels and reducing transfusion dependence in patients with CDA will be determined by the results of ongoing clinical trials. The findings will provide valuable insights into the potential of these agents as a standard treatment option for patients suffering from this condition.
Figure 1 effectively breaks down the complex supportive care strategies for CDA into easily understandable components, highlighting the various treatment approaches, their mechanisms, and their outcomes.
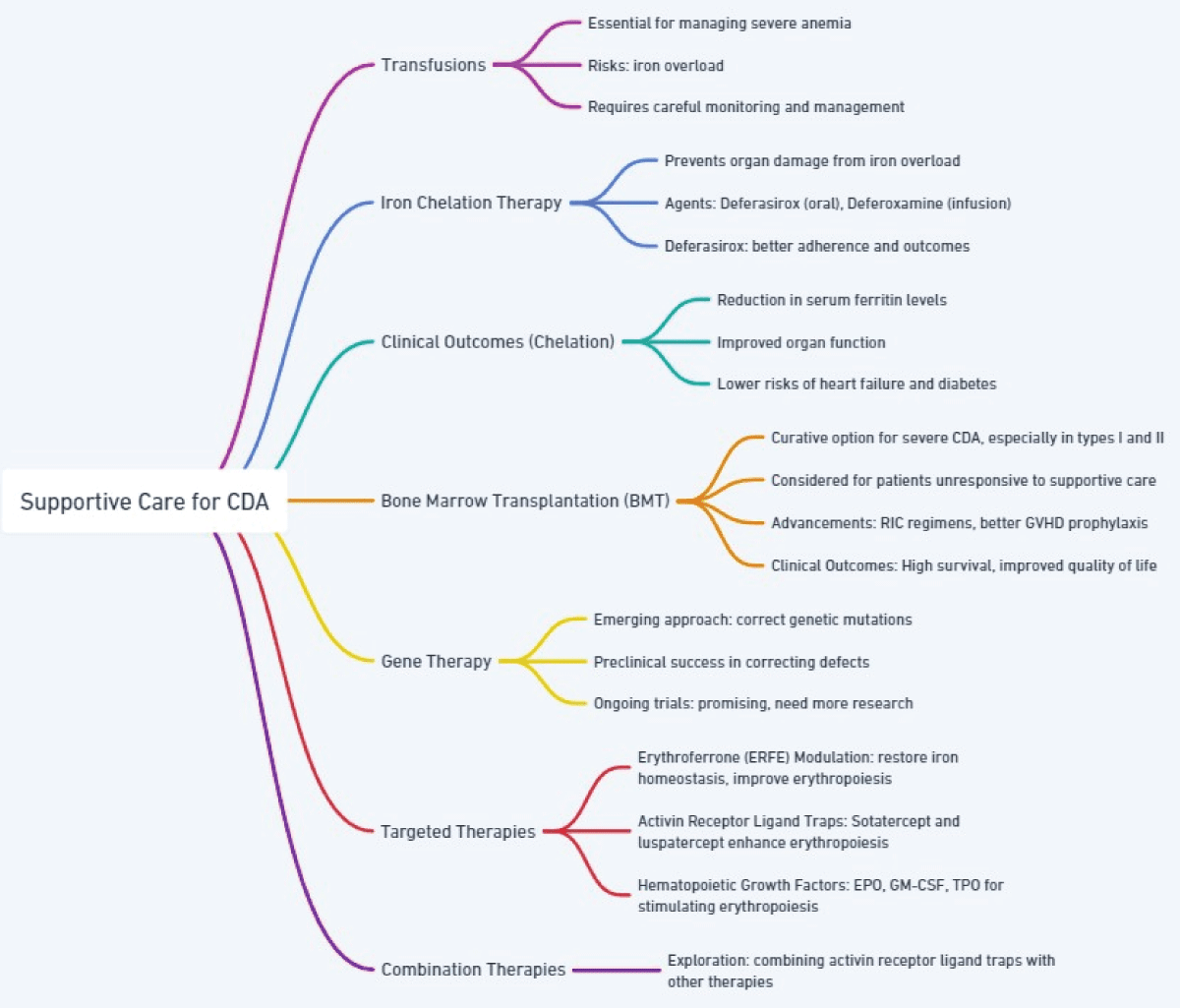
Figure 1: Supportive Care Strategies for Congenital Dyserythropoietic Anemia (CDA).
Perspectives
The treatment of Congenital Dyserythropoietic Anaemias (CDAs) presents a singular challenge, demanding a delicate balance between the management of symptoms and the addressing of the underlying pathophysiology. It is our view, as clinicians, that while recent advancements in therapies such as bone marrow transplantation, gene therapy, and targeted treatments represent significant progress, they also underscore the necessity for personalised approaches. Our clinical experience indicates that it is essential to adapt interventions to the specific characteristics of each patient, considering factors such as the severity of the disease, the genetic subtype, and the potential complications. Furthermore, we acknowledge the significance of prompt diagnosis and multidisciplinary care in enhancing outcomes. While innovative therapies present a promising avenue for treatment, their accessibility remains constrained, particularly in resource-limited settings. This highlights the necessity for global initiatives to reduce disparities in healthcare. Additionally, as novel therapies emerge, we emphasise the necessity for rigorous long-term studies to evaluate their efficacy, safety, and impact on quality of life. This perspective underscores our commitment to advancing CDA management through both clinical innovation and equitable healthcare delivery.
The management of patients with Congenital Dyserythropoietic Anemias (CDA) requires a multidisciplinary team approach due to the complexity of this disease. While supportive care continues to represent a fundamental component of the approach, advances in the fields of bone marrow transplantation, gene therapy, and targeted therapies provide substantial grounds for optimism in terms of the potential for improved patient outcomes. The continuation of research and clinical trials is essential to further elucidate the pathophysiology of CDAs and to develop innovative treatment strategies that can enhance the quality of life for affected individuals. As the understanding of these disorders continues to evolve, the potential for more effective and personalized therapeutic interventions increases, offering patients with CDAs a brighter future.
Declaration
SNOMED CT (Systematized Nomenclature of Medicine-Clinical Terms), UpToDate, PubMed, Cochrane Library, Medscape, Micromedex, DynaMed, and VisualDx were used as cross-reference tools.
- Gambale A, Iolascon A, Andolfo I, Russo R. Diagnosis and management of congenital dyserythropoietic anemias. Expert review of hematology. 2016;9(3):283-296. Available from: https://doi.org/10.1586/17474086.2016.1131608
- Russo R, Iolascon A, Andolfo I, Marra R, Rosato BE. Updates on clinical and laboratory aspects of hereditary dyserythropoietic anemias. International Journal of Laboratory Hematology. 2024. Available from: https://doi.org/10.1111/ijlh.14307
- Iolascon A, Esposito MR, Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. haematologica. 2012;97(12):1786. Available from: https://doi.org/10.3324/haematol.2012.072207
- Renella R, Wood WG. The congenital dyserythropoietic anemias. Hematology/Oncology Clinics. 2009;23(2):283-306. Available from: https://doi.org/10.1016/j.hoc.2009.01.010
- Cazzola M. Ineffective erythropoiesis and its treatment. Blood, The Journal of the American Society of Hematology. 2022;139(16):2460-2470. Available from:https://doi.org/10.1182/blood.2021011045
- Cappellini MD. Exjade®(deferasirox, ICL670) in the treatment of chronic iron overload associated with blood transfusion. Therapeutics and clinical risk management. 2007;3(2):291-299. Available from: https://doi.org/10.2147/tcrm.2007.3.2.291
- Coates TD. Iron overload in transfusion-dependent patients. Hematology 2014, the American Society of Hematology Education Program Book. 2019;2019(1):337-344. Available from: https://doi.org/10.1182/hematology.2019000036
- Wanless IR, Sweeney G, Dhillon AP, Guido M, Piga A, Galanello R, et al. Lack of progressive hepatic fibrosis during long-term therapy with deferiprone in subjects with transfusion-dependent beta-thalassemia. Blood, the Journal of the American Society of Hematology. 2002;100(5):1566-1569. Available from: https://doi.org/10.1182/blood-2002-01-0306
- Maggio A. Light and shadows in the iron chelation treatment of haematological diseases. British journal of haematology. 2007;138(4):407-421. Available from: https://doi.org/10.1111/j.1365-2141.2007.06666.x
- Uygun V, Russo R, Karasu G, Daloğlu H, Iolascon A, Yeşilipek A. Hematopoietic stem cell transplantation in congenital dyserythropetic anemia type II: a case report and review of the literature. Journal of Pediatric Hematology/Oncology. 2020;42(6):e507-510. Available from: https://doi.org/10.1097/mph.0000000000001612
- Braun M, Wölfl M, Wiegering V, Winkler B, Ertan K, Bald R, et al. Successful treatment of an infant with CDA type II by intrauterine transfusions and postnatal stem cell transplantation. Pediatric Blood & Cancer. 2014;61(4):743-745. Available from: https://doi.org/10.1002/pbc.24786
- Gupta V, Braun TM, Chowdhury M, Tewari M, Choi SW. A systematic review of machine learning techniques in hematopoietic stem cell transplantation (HSCT). Sensors. 2020;20(21):6100. Available from: https://doi.org/10.3390/s20216100
- Rangarajan HG, Stanek JR, Abdel-Azim H, Modi A, Haight A, McKinney CM, et al. Hematopoietic cell transplantation for congenital dyserythropoietic anemia: a report from the pediatric transplant and cellular therapy Consortium. Transplantation and Cellular Therapy. 2022;28(6):329-e1. Available from: https://doi.org/10.1016/j.jtct.2022.03.007
- Miano M, Eikema DJ, Aljurf M, van’t Veer PJ, Öztürk G, Wölfl M, et al. Stem cell transplantation for congenital dyserythropoietic anemia: an analysis from the European Society for Blood and Marrow Transplantation. haematologica. 2019;104(8):e335. Available from: https://doi.org/10.3324/haematol.2018.206623
- Sayed N, Allawadhi P, Khurana A, Singh V, Navik U, Pasumarthi SK, et al. Gene therapy: Comprehensive overview and therapeutic applications. Life sciences. 2022;294:120375. Available from: https://doi.org/10.1016/j.lfs.2022.120375
- Dessy-Rodriguez M, Fañanas-Baquero S, Venturi V, Payán-Pernía S, Tornador C, Hernandez G, et al. Modelling Congenital Dyserythropoietic Anemia Type II through Gene Editing in Hematopoietic Stem and Progenitor Cells. Blood. 2020;136:27. Available from: https://doi.org/10.1182/blood-2020-139207
- Kumari R, Kaźmierczak P. Modeling congenital dyserythropoietic anemia in genetically modified mice. Acta Haematologica Polonica. 2022;53(1):26-38. Available from: https://doi.org/10.5603/AHP.a2022.0003
- Khoriaty R, Weyand A, Hesketh G, Bernard A, Everett L, Zhu G, et al. Overlap of SEC23A and SEC23B function suggests a novel therapeutic approach for congenital dyserythropoietic anemia type II. Blood. 2017;130:80. Available from: http://dx.doi.org/10.1182/blood.V130.Suppl_1.80.80
- Chen Y, Wen R, Yang Z, Chen Z. Genome editing using CRISPR/Cas9 to treat hereditary hematological disorders. Gene Therapy. 2022;29(5):207-216. Available from: https://doi.org/10.1038/s41434-021-00247-9
- Dessy-Rodriguez M, Fañanas-Baquero S, Venturi V, Payan S, Tornador C, Hernández G, et al. Lentiviral Gene Therapy for the Correction of Congenital Dyserythropoietic Anemia Type II. Blood. 2021;138:1994. Available from: http://dx.doi.org/10.1182/blood-2021-152332
- Rahimmanesh I, Boshtam M, Kouhpayeh S, Khanahmad H, Dabiri A, Ahangarzadeh S, et al. Gene editing-based technologies for beta-hemoglobinopathies treatment. Biology. 2022;11(6):862. Available from: https://doi.org/10.3390/biology11060862
- Srole DN, Ganz T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. Journal of cellular physiology. 2021;236(7):4888-4901. Available from: https://doi.org/10.1002/jcp.30247
- Babitt JL. Erythroferrone in iron regulation and beyond. Blood, The Journal of the American Society of Hematology. 2022;139(3):319-321. Available from: https://doi.org/10.1182/blood.2021014326
- Koury MJ. Erythroferrone: a missing link in iron regulation. The Hematologist. 2015;12(1).
- Babar S, Saboor M. Erythroferrone in focus: emerging perspectives in iron metabolism and hematopathologies. Blood Science. 2024;6(4):e00198. Available from: https://doi.org/10.1097/bs9.0000000000000198
- van Vuren AJ, Eisenga MF, van Straaten S, Glenthøj A, Gaillard CA, Bakker SJ, et al. Interplay of erythropoietin, fibroblast growth factor 23, and erythroferrone in patients with hereditary hemolytic anemia. Blood advances. 2020;4(8):1678-1682. Available from: https://doi.org/10.1182/bloodadvances.2020001595
- De Rosa G, Andolfo I, Marra R, Manna F, Rosato BE, Iolascon A, et al. RAP-011 rescues the disease phenotype in a cellular model of congenital dyserythropoietic anemia type II by inhibiting the SMAD2-3 pathway. International journal of molecular sciences. 2020;21(15):5577. Available from: https://doi.org/10.3390/ijms21155577
- Lan Z, Lv Z, Zuo W, Xiao Y. From bench to bedside: The promise of sotatercept in hematologic disorders. Biomedicine & Pharmacotherapy. 2023;165:115239. Available from: https://doi.org/10.1016/j.biopha.2023.115239
- Davies AT, Devlin PM, Dugan C, Richards T, Miles LF. Non‐erythropoiesis stimulating agent, non‐iron therapies for the management of anemia: A scoping review. Transfusion. 2023;63(4):849-860. Available from: https://doi.org/10.1111/trf.17274
- Iolascon A, Rivella S, Anagnou NP, Camaschella C, Swinkels D, Muckenthaler MU, Porto G, Barcellini W, Andolfo I, Risitano AM, Kattamis A. The EHA research roadmap: anemias. Hemasphere. 2021;5(7):e607. Available from: https://doi.org/10.1097/hs9.0000000000000607
- Kubasch AS, Fenaux P, Platzbecker U. Development of luspatercept to treat ineffective erythropoiesis. Blood advances. 2021;5(5):1565-1575. Available from: https://doi.org/10.1182/bloodadvances.2020002177
- Lee D, Kim DW, Cho JY. Role of growth factors in hematopoietic stem cell niche. Cell biology and toxicology. 2020;36(2):131-144. Available from: https://doi.org/10.1007/s10565-019-09510-7
- Mann Z, Sengar M, Verma YK, Rajalingam R, Raghav PK. Hematopoietic stem cell factors: their functional role in self-renewal and clinical aspects. Frontiers in Cell and Developmental Biology. 2022;10:664261. Available from: https://doi.org/10.3389/fcell.2022.664261
- Cappellini MD, Marcon A, Fattizzo B, Motta I. Innovative treatments for rare anemias. HemaSphere. 2021 Jun 1;5(6):e576. Available from: https://doi.org/10.1097/hs9.0000000000000576
- Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408-414. Available from: https://doi.org/10.1038/nm.3512
- Carrancio S, Markovics J, Wong P, Leisten J, Castiglioni P, Groza MC, et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br J Haematol. 2014;165(6):870-882. Available from: https://doi.org/10.1111/bjh.12838