
More Information
Submitted: November 22, 2024 | Approved: December 02, 2024 | Published: December 03, 2024
How to cite this article: García-Donas G, Vargas MT, Martínez-Chinchilla C, Alkadi N, Rodríguez A. Congenital Dysfibrinogenaemia: A Family Case Report J Hematol Clin Res. 2024; 8(1): 034-038. Available from:
https://dx.doi.org/10.29328/journal.jhcr.1001032
DOI: 10.29328/journal.jhcr.1001032
Copyright License: © 2024 García-Donas G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Congenital fibrinogen disorders; Dysfibrinogenaemia; Genotype-phenotype correlation; Next-generation sequencing

Congenital Dysfibrinogenaemia: A Family Case Report
G García-Donas1*, MT Vargas2, C Martínez-Chinchilla1, N Alkadi1,3 and A Rodríguez1
1Hematology Department, Virgen Macarena University Hospital, Sevilla, Andalucía, Spain
2Hematology Department, Virgen del Rocío University Hospital. Institute of Biomedicine of Seville (IBIS), Sevilla, Andalucía, Spain
3Technical Advisor, Andalusian Health Service (SAS), Spain
*Address for Correspondence: G García-Donas, Hematology Department, Virgen Macarena University Hospital, Sevilla, Andalucía, Spain, Email: gloriagdonas@hotmail.com; gloria.garciadonas.sspa@juntadeandalucia.es
Congenital fibrinogen qualitative disorders, including dysfibrinogenemia and hypo-dysfibrinogenaemia, are highly heterogeneous, both in clinical manifestation and for the mutational molecular spectrum driving these disorders. Correlations between phenotype and genotype remain poorly defined. Considerable work lies ahead in order to achieve diagnostic and prognostic precision and subsequently provide targeted management for this rare disease. Here we report the laboratory test, the clinical and molecular characterisation of a family with dysfibrinogenemia.
Congenital Fibrinogen Disorders (CFDs) are rare inherited disorders affecting fibrinogen quantity and function [1,2]. Quantitative fibrinogen deficiencies include afibrinogenaemia (absent plasma fibrinogen antigen levels) and hypofibrinogenaemia (proportional decrease of functional and antigenic fibrinogen levels). Qualitative disorders include dysfibrinogenaemia (decreased functional and normal antigenic fibrinogen levels) and hypodysfibrinogenaemia (discrepant decrease of functional and antigenic fibrinogen level, suggested activity: antigen ratio < 0.7) [3,4]. The diagnosis of CFDs is based on a coagulation test with fibrinogen antigen often performed by immunological methods, and activity assays (Clauss fibrinogen), in addition to Thrombin Time (TT) and Reptilase Time (RT) [5]. Genetic mutations in the coding region of one of the three fibrinogen genes, FGA, FGB, or FGG lead to these fibrinogen disorders [6]. Abnormalities in the FGA and FGG genes occur more commonly than in FGB. There are more than 1000 mutations published according to cases listed in the Groupe d’Etude sur l’Hemostase et la Thrombose (GEHT) Human Fibrinogen Database (HFD) [7].
Congenital Disfibrinogenaemia (CD) is the most frequent type of CFD, but its exact prevalence is difficult to establish because of the large number of unreported asymptomatic cases. Approximately 50% - 70% of CD patients are diagnosed incidentally, either during routine laboratory testing or before surgery [8,9]. Clinical presentation in CD is highly variable, from no manifestations to bleeding and/or thrombotic events. However, phenotypic variability and lack of correlation between laboratory values, symptoms manifestation, and disease severity complicate diagnosis and consequently, clinical management [10].
CD is generally associated with autosomal dominant inheritance caused by heterozygosity for missense mutations. The two most frequently mutated hotspots occur at FGA Arg35 and FGG Arg301 [11]. Most cases carrying these hotspot mutations are asymptomatic as detailed on the GEHT HFD database, not significantly associated with either the risk of thrombotic events or with the risk of major bleeding or with pregnancy complications [12-14]. Advances in molecular profiling techniques over the last decade have significantly increased the numbers of published variants in the FGA, FGB, and FGG genes driving these disorders. Few genotypes are clearly correlated with a clinical phenotype [15]. The only description of a rare variant is not sufficient for a correct genetic diagnosis, and the pathogenic role of a point variant should be demonstrated by experimental validation [16]. The correlation between the clinical phenotype and genotype of these patients is usually unclear [17,18]. We describe a family case with dysfibrinogenaemia in order to clarify the genotype–phenotype relationship.
Aims
Determine clinical phenotype, laboratory features, and genotype in a family with CD identified at our centre.
A 56 years old female patient with a history of hypertension and hypercholesterolemia suffered an episode of decreased visual acuity in the right eye in 2021 because of Central Retinal Vein Occlusion (CRVO) so she was diagnosed with CD because of a thrombophilia test. The laboratory test showed reduced Clauss Fibrinogen (CF) with normal Quantitative Fibrinogen (QF). Rotational-thromboelastometry (ROTEM) was assessed to complete functional study and diagnosis was confirmed by genotype. The patient had no history of thrombosis or bleeding despite multiple major surgical interventions. We studied the family, so relatives with CD were clinically evaluated. Samples were analysed using Sysmex coagulometer CS-2500 reagents (PT- Tromborel, APTT- Actin FS, TT- Thromboclotin, RT-Batroxobin, CF-Thrombin). QF with Nephelometry reagent Siemens N Antiserum to Human fibrinogen. The genetic study was an exome analysis using the Next Generation Sequencing (NGS) NEXTseq 1000 sequencing system (Illumina).
The results of the coagulation assays are summarised in Table 1. ROTEM showed basically prolonged Coagulation Time (CT) in EXTEM and minimal shortening in maximum clot firmness (MCF) in FIBTEM (Figure 1). Seven of ten relatives were diagnosed with CD and all of them were asymptomatic. Highlights two sisters with cardiovascular risk factors and no haemostatic complications in multiple major surgeries. The genotype in all CD subjects was variant FGG c.902G>A p. (Arg301His), previously reported [19], in an autosomal dominant pattern. The mutations considered pathogenic were compared against previously described mutations as reported by ClinVar and the HGMD genomic variant classification database. The family also had antecedents of a consanguineous marriage.
| Table 1 | |||||||
| Age (years) at diagnosis | PTr (0,8-1,2) |
APTTr (0,8-1,2) |
TT s (15-20) |
RT s (16-22) |
CF mg/dl (180-350) | QF mg/dl (180-350) | |
| Propositus | 56 | 1,1 | 0,97 | 34,2 | 37,8 | 71,8 | 321 |
| Daughter 1 | 25 | 1,1 | 1,16 | 37,2 | 38,1 | 72 | 324 |
| Daughter 2 | 27 | 1,1 | 1,15 | 35,4 | 38,3 | 90 | 323 |
| Brother 1 | 64 | 1,0 | 1,07 | 18,3 | 21,5 | 270 | NA |
| Sister 1* | 57 | 1,08 | 0,87 | 37,5 | 33,1 | 59,2 | 295 |
| Sister´s 1 son | 20 | 1,18 | 1,0 | 35 | 36 | 58 | 268 |
| Sister´s 1 daughter | 35 | 1,13 | 0,95 | 36 | 35,9 | 63,9 | 227 |
| Sister´s 1 son | 40 | 1,04 | 0,96 | 19 | 23,7 | 294 | NA |
| Sister 2 | 54 | 1,07 | 0,95 | 18,8 | 21,5 | 183 | NA |
| Sister 3** | 67 | 1,16 | 0,98 | 37,1 | 36,6 | 67,6 | 246 |
| Sister´s 3 daughter | 42 | 1,02 | 0,91 | 33,5 | 35,2 | 87 | 372 |
| PT: Prothrombin Time; APTT: Activated Partial Thromboplastin Time; TT: Thrombin Time; TR: Reptilase Time; CF: Clauss Fibrinogen; QF: Quantitative Fibrinogen; NA: Not Aplicable; Shaded in Yellow Affected Relatives; *Diabetes Mellitus (DM); **DM, Dyslipidemia, Smoker, Polio disease | |||||||
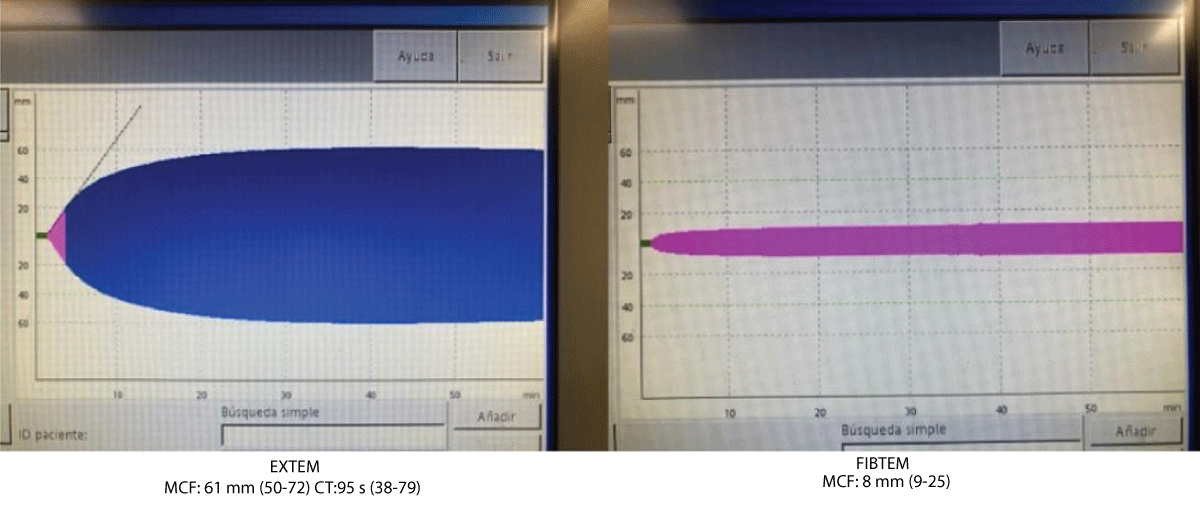
Figure 1: Rotational-Thromboelastometry.
The clinical presentation of fibrinogen disorders does not show a consistent correlation with fibrinogen levels, functional assays, or genotype. The CD is the most frequent type of CFD characterized by dysfunctional fibrinogen (decreased CF) despite normal antigen levels. Various methods of assessing the functional impact are used as viscoelastic assays (thromboelastography or TEG and ROTEM), other less widespread techniques are silico molecular modelling, fibrinolysis assays (e.g. fibrinopeptide release and turbidimetry), and structural studies (e.g. electron microscopy) [18,20,21]. A better understanding of the genetic mechanisms behind this condition is useful for refining diagnostic techniques and treatment strategies according to genotype-phenotype correlation. Some mutations lead to no significant clinical manifestations, while others result in bleeding or thrombosis [22-24]. NGS have moved to the forefront of diagnostic molecular genetic pathology, bypassing traditional Sanger sequencing in their efficiency and rapidity of sequencing ability [25]. However, microarray or multiplex ligation-dependent probe amplification (MLPA) may be helpful if a deletion is suspected because structural variants can be missed with NGS, including large insertions or deletions [26-31].
CD mutations are often inherited in an autosomal dominant manner and are commonly found in exon 2 of FGA (encodes the thrombin cleavage site resulting in knob A exposure) or in exon 8 of FGG (which includes residues encoding hole A and residues necessary for D:D end-to-end assembly of fibrin) [32,33]. Frequencies of both site mutations reach up to 80% - 90% [13,34].
Management of CD remains challenging due to the variable clinical presentation. The majority of patients are asymptomatic or may experience episodes of thrombosis, a hemorrhagic phenotype is reported in some cases. A cause for thrombosis in patients with a CD (for example Fibrinogen Naples) is secondary to elevated levels of thrombin due to reduced sequestration of thrombin by fibrin-thrombin binding [28,35]. The question of whether interacting polymorphisms and variants in other genes known to affect haemostasis and thrombosis lead to a specific phenotype is yet unanswered. There are cases of polymorphisms in the fibrinogen genes and other genes that have been demonstrated to affect plasma fibrinogen levels in Genome-Wide Association Studies (GWAS) [36-38].
Treatment options for thrombosis are anticoagulant medications for a duration that depends on environmental circumstances and thromboprophylaxis in prothrombotic situations. Treatment for bleeding and prophylaxis includes varying types of fibrinogen replacement and the use of other haemostatic agents such as tranexamic acid [39].
The detection of CD in our case is incidental, there is not enough evidence to suggest an association with CRVO, even more so considering that all CD relatives are asymptomatic and taking into account that as previously reported CD because of the variant FGG c.902G>A p. (Arg301His) is mostly casual and asymptomatic, but there are thrombotic and bleeding cases reported [11]. Environmental risk factors must be considered simultaneously as responsible for the thrombotic event. The CD is underdiagnosed for the usual lack of symptoms and normal basic coagulation test. PT and APTT are usually normal but sensitive depending on the coagulometers and activators used [8,25]. It would be useful to better estimate the prevalence of CD to include CF in the basic coagulation test. The TT and RT time is frequently prolonged as is seen in all our CD cases but may differ or be normal range according to the method used [3]. Currently, there is a lack of evidence-based recommendations and consensus on the therapeutic management of (hypo-dysfibrinogenemia) [40]. Therapeutic strategies are mainly based on personal and family bleeding history, thrombosis, and/or pregnancy complications [41]. Genotype–phenotype correlations would be of help to better estimate the risk of these patients. Some dysfibrinogenemia variants are strongly associated with increased susceptibility to thrombosis but other specific variants and the bleeding tendency are not so well established [42]. Global hemostatic assay such as ROTEM would be a help for diagnosis and therapy guidance, especially in case of major surgery, in the propositus case this basal test was minimally altered. Further studies are needed to define whether these tests could be used to monitor the fibrinogen replacement or whether they could be of diagnosis or prognostic value [43].
Dysfibrinogenaemia presents a diagnostic challenge because most patients are asymptomatic and the routine coagulation tests are usually normal. Including CF at the basic coagulation test, could help to get an earlier diagnosis. Robust genotype-phenotype correlations are difficult to establish for fibrinogen disorders. According to the literature, the variant FGG c.902G>A p. (Arg301His), is broadly incidental and asymptomatic. There is no clear link to the thrombotic events so far. Functional studies such as Rotational-thromboelastometry could aid in approach diagnosis and treatment guidance, but the role is not so well established. NGS is useful to support the diagnosis of rare diseases. Further research is needed to deepen the knowledge of the genotype and function of molecular variants with clinical correlation for risk assessment and tailored treatment in these patients.
Ethical considerations
The reporting of clinical and genetic data was approved by the patient and their relatives.
Author contributions
GGD conceived, designed the review, and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
- Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood. 2015 26;125(13):2052-61. Available from: https://doi.org/10.1182/blood-2014-08-532820
- Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, et al. BCSH Committee. Guideline for the diagnosis and management of the rare coagulation disorders: a United Kingdom Haemophilia Centre Doctors' Organization guideline on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2014 ;167(3):304-26. Available from: https://doi.org/10.1111/bjh.13058
- Casini A, Undas A, Palla R, Thachil J, de Moerloose P. Diagnosis and classification of congenital fibrinogen disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2018;16(9):1887–90. Available from: https://doi.org/10.1111/jth.14216
- Cunningham MT, Brandt JT, Laposata M, Olson JD. Laboratory diagnosis of dysfibrinogenemia. Arch Pathol Lab Med. 2002;126(4):499-505. Available from: https://doi.org/10.5858/2002-126-0499-ldod
- Miesbach W, Schenk J, Alesci S, Lindhoff-Last E. Comparison of the fibrinogen Clauss assay and the fibrinogen PT-derived method in patients with dysfibrinogenemia. Thromb Res. 2010;126(6):e428–33. Available from: https://doi.org/10.1016/j.thromres.2010.09.004
- Galanakis DK. Afibrinogenemias and dysfibrinogenemias. In: Marder V, White GC, Aird WC, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Philadelphia: Lippincott Williams & Wilkins. 2012;693-708.
- Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, et al. Ensembl 2022. Nucleic Acids Res. 2022;50(D1):D988–95. Available from: https://doi.org/10.1093/nar/gkab1049
- Shapiro SE, Phillips E, Manning RA, Morse CV, Murden SL, Laffan MA, Mumford AD. Clinical phenotype, laboratory features, and genotype of 35 patients with heritable dysfibrinogenaemia. Br J Haematol. 2013;160(2):220-7. Available from: https://doi.org/10.1111/bjh.12085
- Ebert RF. Index of Variant Human Fibrinogens. Boca Raton, Ann Arbor, Boston: CRC Press; 1994. Available from: https://www.routledge.com/Index-of-Variant-Human-Fibrinogens/Ebert/p/book/9780849389986?srsltid=AfmBOopqoE9HdkcaIiZ7BrhTrATa5hagIOZ2AJ3ShfalPEizfz5JcfOY
- Smith N, Bornikova L, Noetzli L, Guglielmone H, Minoldo S, Backos DS, et al. Identification and characterization of novel mutations implicated in congenital fibrinogen disorders. Res Pract Thromb Haemost. 2018;2(4):800-811. Available from: https://doi.org/10.1002/rth2.12127
- Casini A, Neerman-Arbez M, Ariëns RA, de Moerloose P. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. J Thromb Haemost. 2015;13(6):909-19. Available from: https://doi.org/10.1111/jth.12916
- Neerman-Arbez M, de Moerloose P, Bridel C, Honsberger A, Schönbörner A, Rossier C, et al. Mutations in the fibrinogen α gene account for the majority of cases of congenital afibrinogenemia. Blood. 2000;96(1):149-52. PMID: 10891444. Available from: https://pubmed.ncbi.nlm.nih.gov/10891444/
- Hanss M, Biot F. A database for human fibrinogen variants. Ann NY Acad Sci. 2001;936:89–90. Available from: https://doi.org/10.1111/j.1749-6632.2001.tb03495.x
- Casini A, Blondon M, Lebreton A, Koegel J, Tintillier V, de Maistre E, et al. Natural history of patients with congenital dysfibrinogenemia. Blood. 2015;125(3):553–61. Available from: https://doi.org/10.1182/blood-2014-06-582866
- Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost. 1995;73(1):151-61. PMID: 7740487. Available from: https://pubmed.ncbi.nlm.nih.gov/7740487/
- Asselta R, Platè M, Robusto M, Borhany M, Guella I, Soldà G, et al. Clinical and molecular characterisation of 21 patients affected by quantitative fibrinogen deficiency. Thromb Haemost. 2015;113(3):567-76. Available from: https://doi.org/10.1160/th14-07-0629
- Bor MV, Feddersen S, Pedersen IS, Sidelmann JJ, Kristensen SR. Dysfibrinogenemia-Potential Impact of Genotype on Thrombosis or Bleeding. Semin Thromb Hemost. 2022;48(2):161-173. Available from: https://doi.org/10.1055/s-0041-1730358
- Ramanan R, McFadyen JD, Perkins AC, Tran HA. Congenital fibrinogen disorders: Strengthening genotype-phenotype correlations through novel genetic diagnostic tools. Br J Haematol. 2023;203(3):355-368. Available from: https://doi.org/10.1111/bjh.19039
- Peyvandi F, Kaufman RJ, Seligsohn U, Salomon O, Bolton-Maggs PH, et al. Rare bleeding disorders. Haemophilia. 2006;12 Suppl 3:137-42. Available from: https://doi.org/10.1111/j.1365-2516.2006.01271.x
- Duric N, Szakmany T. The role of rotational thromboelastometry in understanding the coagulation problems in COVID-19 associated critical illness. Anaesthesiol Intensive Ther. 2021;53(4):336-342. Available from: https://doi.org/10.5114/ait.2021.109401
- Reardon B, Pasalic L, Favaloro EJ. The Role of Viscoelastic Testing in Assessing Hemostasis: A Challenge to Standard Laboratory Assays? J Clin Med. 2024;13(12):3612. Available from: https://doi.org/10.3390/jcm13123612
- Williams M, Green K. Role of mass spectrometry in the diagnosis of congenital fibrinogen defects. Blood Coagulation & Fibrinolysis. 2021;32(4):333-338.
- Tian D, Liang J, Gao H, Xu X, Nie W, Yin M, et al. Clinical phenotype and laboratory characteristics of 93 patients with congenital fibrinogen disorders from unrelated 36 families. Res Pract Thromb Haemost. 2024;8:e102445. Available from: https://doi.org/10.1016/j.rpth.2024.102445
- Castaman G, Kessler CM. Genotype-phenotype correlation in congenital dysfibrinogenemia: New insights. Blood Adv. 2022;6(16):4978-4984.
- Paraboschi EM, Duga S, Asselta R. Fibrinogen as a pleiotropic protein causing human diseases: The mutational burden of Aα, Bβ, and γ chains. Int J Mol Sci. 2017;18(12):2711. Available from: https://doi.org/10.3390/ijms18122711
- Zhou J, Ding Q, Chen Y, Ouyang Q, Jiang L, Dai J, et al. Clinical features and molecular basis of 102 Chinese patients with congenital fibrinogen disorders. Blood Cells Mol Dis. 2015;55(4):308–15. Available from: https://doi.org/10.1016/j.bcmd.2015.06.002
- Marchi R, Vilar R, Durual S, Goodyer M, Gay V, Neerman-Arbez M, et al. Fibrin clot properties to assess the bleeding phenotype in unrelated patients with hypodysfibrinogenemia due to novel fibrinogen mutations. Thromb Res. 2021;197:56–64. Available from: https://doi.org/10.1016/j.thromres.2020.11.003
- Ceznerova E, Kaufmanova J, Sovova Ž, Štikarova J, Loužil J, Kotlin R, et al. Structural and functional characterization of four novel fibrinogen mutations in FGB causing congenital fibrinogen disorder. Int J Mol Sci. 2022;23(2):721. Available from: https://doi.org/10.3390/ijms23020721
- Moret A, Zuniga A, Ibanez M, Cid AR, Haya S, Ferrando F, et al. Clinical and molecular characterization by next generation sequencing of Spanish patients affected by congenital deficiencies of fibrinogen. Thromb Res. 2019;180:115–7. Available from: https://doi.org/10.1016/j.thromres.2019.06.015
- Liu X, Wang X, Zhang J. Advances in the genetic diagnosis of congenital dysfibrinogenemia: the role of next-generation sequencing. J Thromb Haemost. 2023;21(7):1355-1363.
- Gindele R, Kerenyi A, Kallai J, Pfliegler G, Schlammadinger A, Szegedi I, et al. Resolving differential diagnostic problems in von Willebrand disease, in fibrinogen disorders, in Prekallikrein deficiency and in hereditary hemorrhagic telangiectasia by next-generation sequencing. Life. 2021;11(3):202. Available from: https://doi.org/10.3390/life11030202
- Neerman-Arbez M, de Moerloose P. Williams Hematology. New York: McGraw-Hill; 2010.
- Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. 2013;121(10):1712-9. Available from: https://doi.org/10.1182/blood-2012-09-306639
- Richard M, Celeny D, Neerman-Arbez M. Mutations accounting for congenital fibrinogen disorders: an update. Semin Thromb Hemost. 2022;48(8):889–903. Available from: https://doi.org/10.1055/s-0041-1742170
- Meh DA, Mosesson MW, Siebenlist KR, Simpson-Haidaris PJ, Brennan SO, DiOrio JP, et al. Fibrinogen Naples I (B beta A68T) nonsubstrate thrombin-binding capacities. Thromb Res. 2001;103(1):63–73. Available from: https://epublications.marquette.edu/biomedsci_fac/47/
- Huffman JE, de Vries PS, Morrison AC, Sabater-Lleal M, Kacprowski T, Auer PL, et al. Rare and low-frequency variants and their association with plasma levels of fibrinogen, FVII, FVIII, and vWF. Blood. 2015;126(11):e19–e29. Available from: https://doi.org/10.1182/blood-2015-02-624551
- de Vries PS, Chasman DI, Sabater-Lleal M, Chen MH, Huffman JE, Steri M, et al. A meta-analysis of 120,246 individuals identifies 18 new loci for fibrinogen concentration. Hum Mol Genet. 2016;25(2):358–70. Available from: https://doi.org/10.1093/hmg/ddv454
- Pankratz N, Wei P, Brody JA, Chen MH, de Vries PS, Huffman JE, et al. Whole-exome sequencing of 14,389 individuals from the ESP and CHARGE consortia identifies novel rare variation associated with hemostatic factors. Hum Mol Genet. 2022;31(18):3120–32. Available from: https://doi.org/10.1093/hmg/ddac100
- Bornikova L, Peyvandi F, Allen G, Bernstein J, Manco-Johnson MJ. Fibrinogen replacement therapy for congenital fibrinogen deficiency. J Thromb Haemost. 2011;9(9):1687-704. Available from: https://doi.org/10.1111/j.1538-7836.2011.04424.x
- Casini A, de Moerloose P. How I treat dysfibrinogenemia. Blood. 2021;138(21):2021–2030. Available from: https://doi.org/10.1182/blood.2020010116
- Li Y, Ding B, Wang X, Ding Q. Congenital (hypo-)dysfibrinogenemia and bleeding: A systematic literature review. Thromb Res. 2022;217:36-47. Available from: https://doi.org/10.1016/j.thromres.2022.07.005
- Casini A, de Moerloose P. Can the phenotype of inherited fibrinogen disorders be predicted? Haemophilia. 2016;22(5):667-75. Available from: https://doi.org/10.1111/hae.12967
- Casini A. From Routine to Research Laboratory: Strategies for the Diagnosis of Congenital Fibrinogen Disorders. Hamostaseologie. 2020;40(4):460-466. Available from: https://doi.org/10.1055/a-1182-3510