
More Information
Submitted: December 31, 2024 | Approved: January 07, 2025 | Published: January 08, 2025
How to cite this article: Makalo L, Adegoke SA, Allen SJ, Kuti BP, Kassama K, Joof S, et al. Clinical Severity of Sickle Cell Anaemia in Children in the Gambia: A Cross-Sectional Study. J Hematol Clin Res. 2025; 9(1): 001-006. Available from:
https://dx.doi.org/10.29328/journal.jhcr.1001033
DOI: 10.29328/journal.jhcr.1001033
Copyright license: © 2025 Makalo L, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Children; Gambia; Severity; Sickle cell disease

Clinical Severity of Sickle Cell Anaemia in Children in the Gambia: A Cross-Sectional Study
Lamin Makalo1,2*, Samuel A Adegoke1-3, Stephen J Allen1,2,4, Bankole P Kuti1-3, Kalipha Kassama5, Sheikh Joof1, Aboulie Camara1, Mamadou Lamin Kijera1 and Egbuna O Obidike1,5
1Department of Paediatrics, Edward Francis Small Teaching Hospital, Banjul, The Gambia
2School of Medicine and Allied Health Sciences, University of The Gambia, The Gambia
3Obafemi Awolowo University, Ile-Ife, Nigeria
4Liverpool School of Tropical Medicine, Liverpool, UK
5Kanifing General Hospital, Kanifing, Kanifing Municipality, The Gambia
6University of Nigeria, Enugu, Nigeria
*Address for Correspondence: Dr. Lamin Makalo, Department of Paediatrics, Edward Francis Small Teaching Hospital, Banjul, The Gambia, Email: [email protected]
Background: Sickle cell anaemia (SCA) in children demonstrates a broad range of clinical manifestations and serious complications. Assessment of disease severity in specific populations is necessary to plan services and optimise care.
Aim: To describe the clinical severity of SCA and associated sociodemographic and clinical factors in children in Gambia.
Methods: The presence of lifetime complications was confirmed by history and review of medical charts. We determined clinical severity using a validated scoring system and related the severity to sociodemographic and clinical factors.
Results: In 130 study participants, ages ranged from 5 to 15 years with a mean (SD) age of 9.74 (2.81) years. Eleven (8.5%) children had had acute chest syndrome, 7 (5.4%) avascular necrosis of the femoral head, 6 (4.6%) gallstones, 5 (3.8%) stroke and 1 (0.8%) priapism over their lifetime. Disease severity was classified as mild in 108 (83.1%) children, moderate in 17 (13.1%) and severe in 5 (3.8%). Age, age at diagnosis, sex, ethnicity, social class, and treatment with hydroxyurea was not significantly correlated with SCA clinical severity (P values 0.10-0.84).
Conclusion: The high proportion of children with mild disease may be due to the high prevalence of Senegalese β-haemoglobin haplotype in the Senegambia subregion. However, the presence of moderate or severe disease in almost 1 in 5 children calls for concerted efforts in SCD care in this region.
Sickle cell anaemia (SCA), the most severe form of sickle cell disorder, is caused by a mutation in the sixth amino acid of the β-globin gene. Children with SCA often manifest symptoms by six months of life, due to the protective effects of foetal haemoglobin (HbF) which has gradually reduced at about this age. However, manifestations could be as early as 4 months of age depending on the level of decline of HbF [1]. These manifestations include vaso-occlusive crises such as dactylitis (hand-foot syndrome), bone pain and abdominal pain crises. Others include chronic haemolysis, increased susceptibility to infections particularly malaria and pneumococcal infections and multi-organ damage [1,2].
SCD has been shown to exhibit great clinical diversity both among patients and within the same person over time [3]. Clinical phenotypes can range from severe manifestations associated with organ dysfunction to just mild or asymptomatic disease [4]. There are periods when patients may enjoy relatively good health, or a “steady state of health” defined as a period free of crisis (pain or haemolytic), infection or any other acute illness for at least four weeks after the last episode of crisis and at least three months after the last blood transfusion [5]. However, some children with SCA require repeated hospitalizations and some present with life-threatening events such as stroke, meningitis, acute chest syndrome, and priapism. The severity of SCA is affected by the amount of haemoglobin F, genetic factors such as HbS haplotype and socio-demographic factors such as socio-economic status and intelligent parenting [6,7].
An important part of the clinical management of SCA is to assess disease severity to identify high-risk cases and institute appropriate monitoring and therapy [8]. There are many SCD scoring systems to assess disease severity but many have limitations when applied in a low-resource setting. The Adegoke and Kuti scoring system is based on simple and largely objective clinical and laboratory parameters, with emphasis on the clinical findings of liver and splenic enlargement as well as the haemogram (Table 1). Advanced neuroimaging such as CT scan or MRI and molecular parameters such as β-globin gene haplotype [9-11] are not included in the scoring system and, therefore, it is applicable in a resource-limited African setting where the disease is common. Although, some other common haemogram parameters like platelet count, or mean corpuscular haemoglobin concentration [12], and biochemical markers like lactate dehydrogenase which have proven correlation with SCD severity are not part of the parameters, this scoring system is still relevant in the study environment. Disease severity can be grouped into mild, moderate and severe with severity groups validated by statistically significant differences in the concentration of foetal haemoglobin [6,13] and has been previously used in children with similar ages and racial backgrounds as in Gambia [14-16].
| Table 1: Severity scoring system by Adegoke and Kuti. | |
| Parameters | Scoring system |
| Number of painful episodes in the previous 12 months, score: | 0 when number of painful episodes is 0 1 when number is 1 2 when number is 2 or 3 3 when number is > 3 |
| Number of transfusions in the previous 12 months, score: | 0 when number of transfusions is 0 1 when number is 1 2 when number is 2 or 3 3 when number is > 3 |
| Number of hospitalisations in the previous 12 months, score: | 0 when number of hospitalisations is 0 1 when number is 1 2 when number is 2 or 3 3 when number is > 3 |
| Liver enlargement, score: | 0 when liver enlargement is < 2 cm 1 when 2 cm to 5 cm 2 when > 5 cm |
| Splenic enlargement, score: | 0 when splenic enlargement is < 5 cm 1 when 5 cm to 10 cm 2 when > 10 cm |
| Packed cell volume, score: | 0 when packed cell volume is ≥ 24% 1 when 18% - 23% 2 when < 18% |
| White blood cell count, score: | 0 when white blood cell count is < 11,000/mm3 1 when between 11,000 and 15,000/mm3 2 when > 15,000/mm3 |
| Lifetime cumulative incidence of specific complications, score: | 5 when CVD is/was present, 0 when absent 3 when ACS is/was present, 0 when absent 3 when pneumococcal meningitis is/was present, 0 when absent 2 when AVN is present, 0 when absent 1 each when gallstone, chronic leg ulcer, osteomyelitis, or priapism is/was present, 0 when absent. |
| CVD: Cerebrovascular Disease; ACS: Acute chest syndrome; AVN: Avascular necrosis | |
Study design
This study was a hospital-based, cross-sectional study.
Study site
The study was carried out at the Paediatric Department of Edward Francis Small Teaching Hospital (EFSTH), Banjul, the only tertiary hospital in The Gambia. The hospital offers both general and specialist paediatric care and receives referrals from all the provinces in the country. The Paediatric Haematology Clinic is held weekly by a Consultant Paediatrician assisted by a Senior Registrar and two Registrars and offers general and specialist care to children with SCD. There are over 300 children with SCD on the clinic register, out of which an average of 30 patients are reviewed weekly. Haematinics, antimalarial and penicillin chemoprophylaxis are prescribed routinely. Some children also receive hydroxyurea (HU). Given that there is a limited supply of HU and many families can not afford to buy the drug, it is provided to children who have required admission to hospital for one or more complications of SCA or those assessed to have a poor quality of life such as being unable to attend school.
Study population
Children aged 5 – 15 years with SCA confirmed by haemoglobin electrophoresis and in steady state who were attending the SCD clinic for a routine follow-up visit. Children whose parents/ caregivers did not give consent were not included in the study.
Sample size calculation: This study is a secondary data analysis of a study of quality of life which recruited 130 children with SCA (Makalo L, et al. Hemoglobin 2024, doi.org/10.1080/03630269. 2024.2440030).
Ethical consideration
Ethical approval for this study was obtained from the Research Ethics Committee of the EFSTH, Banjul, The Gambia, IRB Number EFSTH-REC-2024-002. The investigator explained the purpose and objectives of the study to the parents in a language they understood. Written informed consent was obtained from the participants, with the study’s scope, purpose, and benefits clearly explained to the parents. The identities of all participants were kept confidential.
Data collection
Consecutive children aged 5-15 years with SCA attending clinic and assessed to be in a steady state were recruited by LM. A data proforma was used to obtain information about socio-demographic characteristics such as age at last birthday, sex and socio-economic class of participants based on rank assessment of parental occupation and the highest level of educational attainment as described by Ogunlesi [17]. The scores for the occupation and educational attainment of both parents ranged from 1 to 5 and were added together and divided by 4 to get the final value approximated to the nearest whole number. Socioeconomic class was graded on a score of 1 to 5; 1 being the highest (Class I) and 5 the lowest (Class V). Classes I and II were further classified as upper social class, class III as middle social class, while IV-V were classified as lower social class.
Information on the age at diagnosis of SCA was documented. This was dichotomised into early diagnosis if the diagnosis was made in the first year of life, or late diagnosis if the diagnosis was made after infancy. Information on the use of hydroxyurea was also obtained.
In addition to general physical examination, abdominal examination measured spleen and liver enlargement below the left and right subcostal margins, respectively.
Subsequently, the venepuncture site was disinfected with methylated spirit, and one millilitre of venous blood was collected into the EDTA bottle for haematological indices at the Pediatric laboratory of the hospital. Analysis of the sample was done using ABX micro-ES 60 automated haemoanalyser (Siemens, Vienna Austria) within three hours of blood collection. Haemoglobin genotype SS had already been determined by alkaline cellulose acetate haemoglobin electrophoresis with quality control in place in the same laboratory and recorded in patients’ folders.
Assessment of sickle cell anaemia severity
The severity of SCA was determined based on the number of admissions, blood transfusions and significant painful crises (pain episodes that required hospital visits and the use of analgesics) in the last 12 months. Other parameters included liver and splenic enlargement, packed cell volume, white blood cell count, and cumulative SCD-related complications as described by Adegoke and Kuti [4]. The presence of lifetime complications was confirmed by history and review of medical charts. The complications were defined as follows:
Acute Chest Syndrome (ACS): An acute illness characterized by fever and respiratory symptoms (dyspnoea, chest pain and cough) accompanied by new pulmonary infiltrates on chest radiograph.
Avascular Necrosis (AVN): Osteonecrosis or aseptic necrosis of the head of the femur or humerus confirmed by radiography as irregularity of the articular surfaces of the head of the femur/humerus.
Stroke (cerebrovascular disease): Acute neurologic symptoms and signs lasting more than 24 hours, secondary to occlusion of and or haemorrhage from cerebral vessels. When available, the diagnosis is confirmed on a computerized tomography (CT) scan or magnetic resonance imaging (MRI).
Severe bacterial infections: Pneumonia, sepsis, meningitis, osteomyelitis, and septic arthritis confirmed by positive culture and or radiograph.
Chronic leg ulcer: Ulceration of the skin and underlying tissue of the lower extremities, especially on the medial or lateral surface of the ankle
Cholelithiasis: The presence of gallstone manifested with severe colicky abdominal pain and confirmed with abdominal ultrasonography.
Priapism: Presence of sustained, unwanted, and painful penile erection.
Data analysis
Data were analysed using SPSS for Windows version 22 statistical software (IBM Corp., Armonk, NY, USA). Categorical variables were summarised using percentages and proportions and compared using Pearson’s chi-square test. Continuous variables were summarised using mean and standard deviations after the Kolmogorov-Smirnov test confirmed normal distribution. The exception was age at diagnosis which was described with median and interquartile range (IQR) because it was not normally distributed.
Over seven months (November 2022 to May 2023), a total of 130 children with SCA who routinely attend the clinic were recruited.
Socio-demographic characteristics
Gender, age and social class: There was an overall male preponderance (53.1%) with a male-to-female ratio of 1.1:1 (Table 2).
| Table 2: Sociodemographic characteristics of study participants. | |
| Sociodemographic variables | HbSS group |
| (n = 130) | |
| n (%) | |
| Gender | |
| Male | 69 (53.1) |
| Female | 61 (46.9) |
| Age group (years) | |
| 5 – 7 | 31 (23.8) |
| 8 – 10 | 50 (38.5) |
| 11 – 13 | 35 (26.9) |
| 14 – 15 | 14 (10.8) |
| Ethnicity | |
| Mandinka | 63 (48.5) |
| Wolof | 23 (17.7) |
| Fula | 22 (16.9) |
| Jola | 7 (5.4) |
| Others* | 15 (11.5) |
| Social class: | |
| Class I | 1 (0.8) |
| Class II | 2 (1.5) |
| Class III | 24 (18.5) |
| Class IV | 53 (40.8) |
| Class V | 50 (38.4) |
| *Others: Serahule, 12 (4.6%); Serer, 9 (3.5%); Manjago, 7 (2.7%) and Aku, 6 (2.3%). | |
Age ranged from 5 to 15 years with a mean (SD) age of 9.74 (2.81) years. The majority of the children were in the age group 8 – 10 years) 50; (38.5%) and the most common ethnicity was Mandinka (63; 48.5%).
Only 3 (2.3%) children were classified as upper socioeconomic class (i.e., classes I and II) and the majority were lower socioeconomic class (xx, xx.x%; classes IV and V).
Age at diagnosis of sickle cell anaemia and use of hydroxyurea: The age of diagnosis of SCA ranged from 9 months to 14 years, with a median (IQR) of 3.0 (2.0, 6.0) years. None of the children was diagnosed in the newborn period or before the first 9 months of life. Twenty-four (18.5%) were diagnosed between the ages of 9 and 12 months. The majority, 68 (52.3%), were diagnosed when aged 1 – 5 years and the remaining 38 (29.2%) after the age of five years. Ninety (69.2%) children were using hydroxyurea at the time of recruitment.
Sickle cell disease clinical severity scoring (Table 3)
| Table 3: Burden of sickle cell disease and disease severity among the 130 children with sickle cell anaemia. | ||
| Variables | Number | Percentage (%) |
| Frequency of significant pain episodes* | ||
| No pain episode | 15 | 11.5 |
| 1 | 25 | 19.2 |
| 2 – 3 | 28 | 21.5 |
| >3 | 62 | 47.7 |
| Frequency of hospitalisations* | ||
| No hospitalisation | 68 | 52.3 |
| 1 | 29 | 22.3 |
| 2 – 3 | 19 | 14.6 |
| >3 | 14 | 10.8 |
| Frequency of transfusions* | ||
| No transfusion | 95 | 73.1 |
| 1 | 25 | 19.2 |
| 2 – 3 | 9 | 6.9 |
| >3 | 1 | 0.8 |
| Liver size | ||
| <2 cm | 124 | 95.4 |
| 2 – 5 cm | 4 | 3.1 |
| >5 cm | 2 | 1.5 |
| Spleen size | ||
| <5 cm | 123 | 94.6 |
| 5 – 10 cm | 6 | 4.6 |
| >10 cm | 1 | 0.8 |
| Packed cell volume | ||
| ≥ 24% | 74 | 56.9 |
| 18% – 23% | 52 | 40.0 |
| <18% | 4 | 3.1 |
| White blood cell count | ||
| <11,000/mm3 | 73 | 56.2 |
| 11,000 - 15,000/mm3 | 38 | 29.2 |
| >15,000/ mm3 | 19 | 14.6 |
| SCD severity | ||
| Mild | 108 | 83.1 |
| Moderate | 17 | 13.1 |
| Severe | 5 | 3.8 |
| Key: * Indicate events within the last 12 months before the study period | ||
Parameters in the twelve months before the study period
Table 3 shows that 115 (88.5%) children experienced at least one significant painful crisis in the 12 months preceding the study. Sixty-two (47.7%) had at least one hospital admission, including 14 (10.8%) who were admitted more than three times in the 12 months preceding the study period. Also, 35 (26.9%) were transfused out of which one child (0.8%) was transfused on more than three occasions in the last 12 months.
Clinical and some laboratory parameters at presentation
Two children with SCA (1.5%) had hepatomegaly exceeding 5cm and 7 (5.4%) had splenomegaly of at least 5cm, including a child (0.8%) with massive splenomegaly (spleen size of 12cm). Four (3.1%) children had PCV < 18% and 57 (43.8%) had WBC counts ≥11,000/mm3 (Table 3).
Complications of sickle cell anaemia
A total of 30 (23.1%) children had SCA-related complications (Figure 1). The leading complication was ACS, which had occurred in 11 (8.5%) children. None of the children had a history or case note documented evidence suggestive of meningitis, chronic leg ulcer, osteomyelitis, or nephropathy. No child had more than one complication.
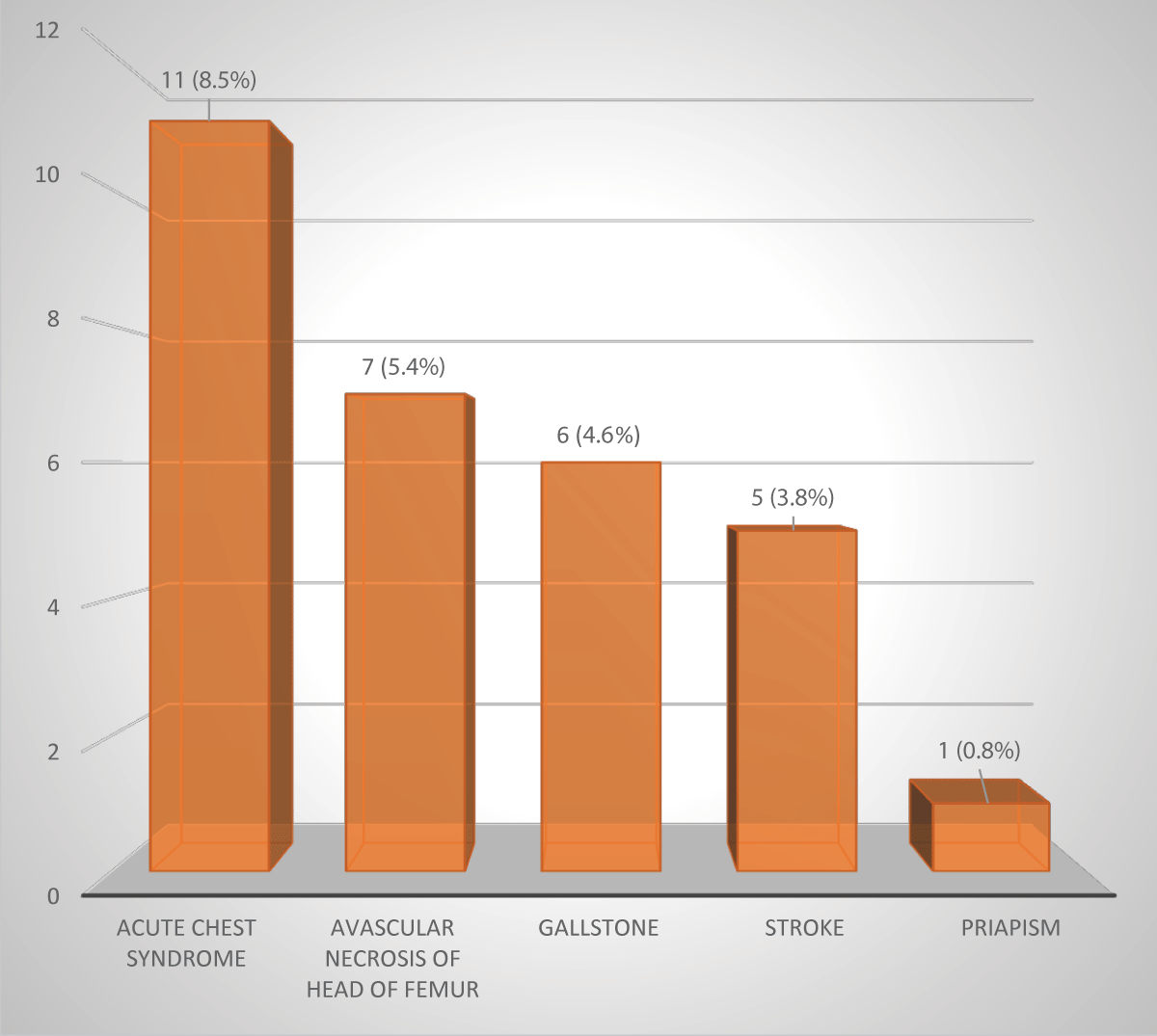
Figure 1: Complications of sickle cell anaemia among the 130 children.
Disease severity scores ranged from 0 - 19 with a mean (SD) score of 5.0 (3.6). One hundred and eight (83.1%) children had mild disease, 17 (13.1%) had moderate disease and 5 (3.8%) had severe disease (Table 3). The latter comprised all five children with stroke who had SCD severity scores of either 18 or 19. Table 4 shows no significant difference in the proportions of patients preadolescents and adolescents who had mild or moderate/severe disease severity. Ditto for gender, social class, timing of diagnosis or the use of hydroxyurea. Also, Table 5 which compared the mean (SD) SCD score between various groups showed no significant difference.
| Table 4: Sociodemographic and Clinical Factors according to disease Severity. | ||||
| Factor | Mild SCD | Moderate and Severe SCD | x2 | P |
| Age Group | ||||
| Preadolescent | 50 | 9 | 0.214 | 0.644 |
| Adolescent | 58 | 13 | ||
| Sex | ||||
| Male | 55 | 14 | 2.190 | 0.164 |
| Female | 53 | 8 | ||
| Ethnicity | ||||
| Mandinka | 52 | 11 | 7.270 | 0.839 |
| Fula | 18 | 4 | ||
| Wollof | 18 | 5 | ||
| Jola | 6 | 1 | ||
| Others | 14 | 1 | ||
| Social Class | ||||
| Upper/Middle | 24 | 3 | 0.380 | 0-538 |
| Lower | 84 | 19 | ||
| Diagnosis | ||||
| Early | 19 | 5 | 0.320 | 0.572 |
| Late | 89 | 17 | ||
| Hydroxyurea Use | ||||
| Yes | 78 | 12 | 2.681 | 0.102 |
| No | 30 | 10 | ||
| Table 5: Mean (SD) sickle cell disease severity score between different groups. | |||
| Variables | Mean (SD) SCD severity score | t-test/ANOVA | p value |
| Age group | 0.534 | 0.594 | |
| Preadolescents | 5.2 (3.8) | ||
| Adolescents | 4.9 (3.5) | ||
| Sex | 0.698 | 0.487 | |
| Male | 5.2 (4.2) | ||
| Female | 4.8 (2.9) | ||
| Social class | 0.322 | 0.748 | |
| Upper social class | 5.7 (4.7) | ||
| Middle/ Lower class | 5.0 (3.6) | ||
| Timing of diagnosis | -0.299 | 0.765 | |
| Early diagnosis | 4.9 (3.2) | ||
| Late diagnosis | 5.1 (3.2) | ||
| Use of hydroxyurea | 1.263 | 0.209 | |
| Yes | 4.7 (3.7) | ||
| No | 5.6 (3.3) | ||
The study revealed a wide range in severity of SCA in children in The Gambia. More than four-fifths of our patients had mild disease, over 17 % had moderate disease, and approximately 3% had severe disease. Overall, our patients tended to have mild disease (severity score < 5) like Senegalese, Yemen and Eastern Saudi Arabia children (average scores 7.8, 5.8, and 4.5, respectively [18,19]. This is unlike the higher mean scores of 9.85 found in Nigerian children and 9.5 reported for children from the southwestern Province of Saudi Arabia [19]. This variability could be linked to the differences in β-globin haplotype polymorphisms in different geographical locations [18]. Saudi and Senegal haplotypes commonly result in mild disease while Benin and Bantu haplotypes found in Nigerian children are generally associated with severe disease [20,21].
In this study, the disease severity score was not correlated with age, (r = 0.2; p = 0.644). This may be attributable to a lower likelihood of lifetime complications in older children with SCA. In addition, gender and socioeconomic class did not influence disease severity scores. This contrasts with findings from Ballas. et al. that showed that pre-adolescent girls and children from lower socioeconomic levels experience more severe illness [21]. Ordinarily, children from higher socioeconomic levels with better education and financial status would be expected to have lower severity scores because of the interplay between genetic and environmental factors on SCA severity. The healthcare system in the Gambia is highly accessible and affordable, unlike many counterparts in the subregion, and this may have reduced the impact of low socioeconomic status.
The diagnosis of SCA was made in the first year of life in only 24 (18.5%) children with SCA, with none of them in the neonatal period or the first 6 months of life. Although age at diagnosis was not associated with disease severity (p = 0.57), early diagnosis is a high priority for improving survival and reducing complications in SCA [22].
Drugs such as hydroxyurea have been associated with a reduction in the severity and frequency of significant painful crises [23]. Although its use was not significantly associated with disease severity in our patients (p = 0.10), we were unable to ascertain compliance with hydroxyurea which may have affected our findings. Besides, treatment with Hydroxyurea in the majority of children might have contributed to the low scores. Especially as HU is prescribed for the more severe cases. Also, only steady-state children were included.
Limitations of our study are that information on the lifetime incidence of complications, previous treatment at other hospitals, and age and symptoms at initial diagnosis may have been subject to recall bias, particularly given that children in steady state were recruited for this study. Other proven parameters of disease severity such as measurement of foetal haemoglobin as well as investigations like cranial magnetic resonance angiography and transcranial Doppler ultrasound scan were not incorporated into the scoring system. Diagnosis of stroke as a complication of sickle cell disease was based on clinical definition, rather than by MRI, which is a limitation since some patients might have been missed without MRI. The inclusion of a more accurate assessment of disease complications facilitated by imaging and other technologies may improve the reliability of the score. Finally, this study did not include an assessment of the number of deaths due to SCA in children in our population. Rather, the study is limited to survivors who likely have less severe disease.
Concurring with Adegoke with Kuti’s conclusion (add ref), further studies employing the most recently described molecular and genetic prognostic markers in SCA are critical in the identification of patients at risk for severe disease and prediction of serious complications of SCA. However, the routine evaluation of disease severity using simple clinical-laboratory parameters is beneficial and may enhance the provision and quality of care and management outcomes for children with this chronic and debilitating disease.
In conclusion, assessment of disease severity for common diseases such as SCA informs the provision of care. Although the majority (83.1%) of children with SCA in this study had mild disease, severe complications occurred in many children. Efforts should be made to strengthen comprehensive SCD care in the country. This should include neonatal screening for early diagnosis, active prevention and management of pain episodes and lifetime complications, especially stroke, to improve disease severity. These strategies should include the continued availability of hydroxyurea and other effective stroke prevention interventions. Periodic assessment of disease severity so that appropriate preventive and therapeutic care could be offered.
We thank the Lecturers/ Consultants in the Department of Paediatrics, EFSTH namely: Prof. Vivian Muoneke, Dr. Kalifa Bojang, Dr. Helen Brotherten, Dr. Simon Pius and Dr. Beatrice Ezenwa for their support and encouragement.
We are grateful for the support and cooperation of medical residents and nurses, especially, Abdoulie Camara in the Department.
- Egesa WI, Nakalema G, Waibi WM, Turyasiima M, Amuje E, Kiconco G, et al. Sickle Cell Disease in Children and Adolescents: A Review of the Historical, Clinical, and Public Health Perspective of Sub-Saharan Africa and Beyond. Int J Pediatr. 2022;2022:3885979. Available from: https://doi.org/10.1155/2022/3885979
- Inusa BPD, Hsu LL, Kohli N, Patel A, Ominu-Evbota K, Anie KA, et al. Sickle Cell Disease-Genetics, Pathophysiology, Clinical Presentation and Treatment. Int J Neonatal Screen. 2019;5(2):20. Available from: https://doi.org/10.3390/ijns5020020
- Farrell AT, Panepinto J, Carroll CP, Darbari DS, Desai AA, King AA, et al. Endpoints for sickle cell disease clinical trials: patient-reported outcomes, pain, and the brain. Blood Adv. 2019;3(23):3982-4001. Available from: https://doi.org/10.1182/bloodadvances.2019000882
- Adegoke SA, Kuti BP. Evaluation of clinical severity of sickle cell anemia in Nigerian children. J Applied Hematol. 2013;4(2):58-64. Available from: https://www.researchgate.net/publication/255710803_Evaluation_of_Clinical_Severity_of_Sickle_Cell_Anaemia_in_Nigerian_Children
- Ballas SK, Lieff S, Benjamin LJ, Dampier CD, Heeney MM, Hoppe C, et al. Investigators, Comprehensive Sickle Cell Centers. Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol. 2010;85(1):6-13. Available from: https://doi.org/10.1002/ajh.21550
- Bitoungui VJ, Pule GD, Hanchard N, Ngogang J, Wonkam A. Beta-globin gene haplotypes among Cameroonians and review of the global distribution: is there a case for a single sickle mutation origin in Africa? OMICS. 2015;19(3):171-9. Available from: https://doi.org/10.1089/omi.2014.0134
- Adeodu OO, Akinlosotu MA, Adegoke SA, Oseni SBA. Foetal Haemoglobin and Disease Severity in Nigerian Children with Sickle Cell Anaemia. Mediterr J Hematol Infect Dis. 2017;9(1):e2017063. Available from: https://doi.org/10.4084/MJHID.2017.063
- Mir R, Sharaf F, FM A. Hemoglobin Subunit Beta Gene Polymorphism rs33949930 T>C and Risk of Sickle Cell Disease—A Case-Control Study from Tabuk (Northwestern Part of Saudi Arabia). Int J Clin Med. 2016;7:25-31. Available from: https://www.scirp.org/journal/paperinformation?paperid=62941
- Ashley-Koch A, Yang Q, Onley RS. Sickle haemoglobin (HbS) Allele and Sickle Cell Disease: a HuGE Review. Am J Epidemiol. 2000;9:839-845. Available from: https://doi.org/10.1093/oxfordjournals.aje.a010288
- Adekile AD, Adeodu OO, Adegoke SA. Haemoglobinopathies. In: Azubuike JC, Nkanginieme KE, Ezechukwu C, Nte AR, Adedoyin AT, editors. Paediatrics and Child Health in a Tropical Region, 3rd edition. Lagos, Nigeria: Educational Printing and Publishing; 2017;1051–1065.
- Serjeant GR. Sickle Cell Disease. Lancet. 1997;350:725-730. Available from: https://doi.org/10.1016/s0140-6736(97)07330-3
- Okocha E, Onwubuya E, Osuji C, Ahaneku G, Okonkwo U. Disease Severity Scores and Haemogram Parameters in Nigerian Sickle Cell Disease Patients. J Blood Disord Transfus. 2015;6:324. Available from: https://www.walshmedicalmedia.com/open-access/disease-severity-scores-and-haemogram-parameters-in-nigerian-sickle-cell-disease-patients-2155-9864-1000324.pdf
- Adeodu OO, Akinlosotu MA, Adegoke SA, Oseni SBA. Foetal haemoglobin and disease severity in Nigerian children with sickle cell anaemia. Mediterr J Hematol Infect Dis. 2017;9(1):e2017063. Available from: https://doi.org/10.4084/MJHID.2017.063
- Adegoke SA, Adeodu OO, Adekile AD. Sickle cell disease clinical phenotypes in children from South-Western Nigeria. Niger J Clin Pract. 2015;18:95-101. Available from: https://doi.org/10.4103/1119-3077.146987
- Adeodu OO, Akinlosotu MA, Adegoke SA, Oseni SBA. Foetal haemoglobin and disease severity in Nigerian children with sickle cell anaemia. Mediterr J Hematol Infect Dis. 2017;9(1):e2017063. Available from: https://doi.org/10.4084/MJHID.2017.063
- Oladimeji OI, Adeodu OO, Onakpoya OH, Adegoke SA. Prevalence of ocular abnormalities in relation to sickle cell disease severity among children in South-western, Nigeria. Eur J Ophthalmol. 2021;31(5):2659-2665. Available from: https://doi.org/10.1177/1120672120957615
- Ogunlesi TA, Dedeke IOF, Kuponiyi OT. Socio-economic classification of children attending Specialist paediatric centres in Ogun State, Nigeria. Niger Med Pract. 2008;54(1):21-25. Available from: https://www.ajol.info/index.php/nmp/article/view/28943
- el-Hazmi MA. Clinical and haematological diversity of sickle cell disease in Saudi children. J Trop Pediatr. 1992;38:106-12. Available from: https://doi.org/10.1093/tropej/38.3.106
- Al-Saqladi AM, Delpisheh A, Bin-Gadeem HA, Brabin BJ. Severity of sickle cell disease in Yemeni children. J Trop Pediatr. 2009;55:208-9. Available from: https://doi.org/10.1093/tropej/fmn109
- Jastaniah W. Epidemiology of sickle cell disease in Saudi Arabia. Ann Saudi Med. 2011;31:289-293. Available from: https://doi.org/10.4103/0256-4947.81540
- Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med. 2001;20:107-21. Available from: https://pubmed.ncbi.nlm.nih.gov/12673836/
- McGann PT. Time to Invest in Sickle Cell Anemia as a Global Health Priority. Pediatrics. 2016;137(6):e20160348. Available from: https://doi.org/10.1542/peds.2016-0348
- Ballas SK, Barton FB, Waclawiw MA, Swerdlow P, Eckman JR, Pegelow CH, et al. Hydroxyurea and sickle cell anemia: effect on quality of life. Health Qual Life Outcomes. 2006;4:59. Available from: https://doi.org/10.1186/1477-7525-4-59